Lecture 9 - Graph Neural Networks#
Learning goals#
Build basic MLP-style Neural Networks from scratch using PyTorch.
Explain molecules as graphs: atoms = nodes, bonds = edges, features at each.
Write the message passing equations and understand neighborhood aggregation.
Build a tiny GNN in PyTorch on toy molecules.
Prepare molecular graphs from SMILES and run a mini GNN.
1. Setup#
We reuse most of the stack from earlier lectures.
Show code cell source
# 1. Setup
import warnings, math, os, sys, json, time, random
warnings.filterwarnings("ignore")
import numpy as np
import pandas as pd
import matplotlib.pyplot as plt
import networkx as nx
# Sklearn bits for splitting and metrics
from sklearn.model_selection import train_test_split
from sklearn.preprocessing import StandardScaler
from sklearn.pipeline import Pipeline
from sklearn.compose import TransformedTargetRegressor
from sklearn.tree import DecisionTreeRegressor
from sklearn.ensemble import RandomForestRegressor
from sklearn.neural_network import MLPRegressor
import copy
from sklearn.metrics import (mean_squared_error, mean_absolute_error, r2_score,
accuracy_score, precision_score, recall_score,
f1_score, roc_auc_score, roc_curve, confusion_matrix)
# Torch for MLP and GNN
try:
import torch
import torch.nn as nn
from torch.utils.data import Dataset, DataLoader
import torch.nn.functional as F
except Exception as e:
print("Installing torch, this may take a minute...")
%pip -q install torch
import torch
import torch.nn as nn
from torch.utils.data import Dataset, DataLoader
# RDKit
try:
from rdkit import Chem
from rdkit.Chem import Draw, Descriptors, Crippen, rdMolDescriptors, AllChem
except Exception:
try:
%pip install rdkit
from rdkit import Chem
from rdkit.Chem import Draw, Descriptors, Crippen, rdMolDescriptors, AllChem
except Exception as e:
print("RDKit is not available in this environment. Drawing and descriptors will be skipped.")
Chem = None
# A small global seed helper
def set_seed(seed=0):
random.seed(seed); np.random.seed(seed); torch.manual_seed(seed)
set_seed(0)
2 Building MLP from scratch#
In Lecture 8 we built MLPs with scikit‑learn. Today we start by doing the same with PyTorch. One advantage of using PyTorch is that it lets you write the forward pass directly, control the training loop, and later move to GPUs or custom layers. We start tiny and stay friendly.
We will predict melting point from four descriptors: MolWt, LogP, TPSA, NumRings, same as before.
2.1 PyTorch regression on melting point#
Every PyTorch project has three parts:
Model: stack of layers with weights and activations.
Loss: a number telling how far predictions are from the truth.
Optimizer: an algorithm that adjusts weights to reduce loss.
We use descriptors [MolWt, LogP, TPSA, NumRings] to predict Melting Point.
url = "https://raw.githubusercontent.com/zzhenglab/ai4chem/main/book/_data/C_H_oxidation_dataset.csv"
df_oxidation_raw = pd.read_csv(url)
def calc_descriptors(smiles):
mol = Chem.MolFromSmiles(smiles)
if mol is None:
return pd.Series({
"MolWt": None,
"LogP": None,
"TPSA": None,
"NumRings": None
})
return pd.Series({
"MolWt": Descriptors.MolWt(mol), # molecular weight
"LogP": Crippen.MolLogP(mol), # octanol-water logP
"TPSA": rdMolDescriptors.CalcTPSA(mol), # topological polar surface area
"NumRings": rdMolDescriptors.CalcNumRings(mol) # number of rings
})
# Apply the function to the SMILES column
desc_df = df_oxidation_raw["SMILES"].apply(calc_descriptors)
# Concatenate new descriptor columns to original DataFrame
df = pd.concat([df_oxidation_raw, desc_df], axis=1)
df
| Compound Name | CAS | SMILES | Solubility_mol_per_L | pKa | Toxicity | Melting Point | Reactivity | Oxidation Site | MolWt | LogP | TPSA | NumRings | |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 0 | 3,4-dihydro-1H-isochromene | 493-05-0 | c1ccc2c(c1)CCOC2 | 0.103906 | 5.80 | non_toxic | 65.8 | 1 | 8,10 | 134.178 | 1.7593 | 9.23 | 2.0 |
| 1 | 9H-fluorene | 86-73-7 | c1ccc2c(c1)Cc1ccccc1-2 | 0.010460 | 5.82 | toxic | 90.0 | 1 | 7 | 166.223 | 3.2578 | 0.00 | 3.0 |
| 2 | 1,2,3,4-tetrahydronaphthalene | 119-64-2 | c1ccc2c(c1)CCCC2 | 0.020589 | 5.74 | toxic | 69.4 | 1 | 7,10 | 132.206 | 2.5654 | 0.00 | 2.0 |
| 3 | ethylbenzene | 100-41-4 | CCc1ccccc1 | 0.048107 | 5.87 | non_toxic | 65.0 | 1 | 1,2 | 106.168 | 2.2490 | 0.00 | 1.0 |
| 4 | cyclohexene | 110-83-8 | C1=CCCCC1 | 0.060688 | 5.66 | non_toxic | 96.4 | 1 | 3,6 | 82.146 | 2.1166 | 0.00 | 1.0 |
| ... | ... | ... | ... | ... | ... | ... | ... | ... | ... | ... | ... | ... | ... |
| 570 | 2-naphthalen-2-ylpropan-2-amine | 90299-04-0 | CC(C)(N)c1ccc2ccccc2c1 | 0.018990 | 10.04 | toxic | 121.5 | -1 | -1 | 185.270 | 3.0336 | 26.02 | 2.0 |
| 571 | 1-bromo-4-(methylamino)anthracene-9,10-dione | 128-93-8 | CNc1ccc(Br)c2c1C(=O)c1ccccc1C2=O | 0.021590 | 7.81 | toxic | 154.0 | -1 | -1 | 316.154 | 3.2662 | 46.17 | 3.0 |
| 572 | 1-[6-(dimethylamino)naphthalen-2-yl]prop-2-en-... | 86636-92-2 | C=CC(=O)c1ccc2cc(N(C)C)ccc2c1 | 0.017866 | 8.58 | toxic | 128.3 | -1 | -1 | 225.291 | 3.2745 | 20.31 | 2.0 |
| 573 | 1,2-dimethoxy-12-methyl-[1,3]benzodioxolo[5,6-... | 34316-15-9 | COc1ccc2c(c[n+](C)c3c4cc5c(cc4ccc23)OCO5)c1OC | 0.016210 | 5.54 | toxic | 215.6 | -1 | -1 | 348.378 | 3.7166 | 40.80 | 5.0 |
| 574 | dimethyl anthracene-1,8-dicarboxylate | 93655-34-6 | COC(=O)c1cccc2cc3cccc(C(=O)OC)c3cc12 | 0.016761 | 5.43 | toxic | 175.3 | -1 | -1 | 294.306 | 3.5662 | 52.60 | 3.0 |
575 rows × 13 columns
df_reg = df[["MolWt", "LogP", "TPSA", "NumRings", "Melting Point"]].dropna()
X = df_reg[["MolWt", "LogP", "TPSA", "NumRings"]].values.astype(np.float32)
y = df_reg["Melting Point"].values.astype(np.float32).reshape(-1, 1)
X_tr, X_te, y_tr, y_te = train_test_split(X, y, test_size=0.2, random_state=42)
scaler = StandardScaler().fit(X_tr)
X_tr_s, X_te_s = scaler.transform(X_tr), scaler.transform(X_te)
X_tr[:3], X_tr_s[:3]
(array([[226.703 , 3.687 , 26.3 , 1. ],
[282.295 , 2.5255, 52.6 , 3. ],
[468.722 , 7.5302, 43.37 , 5. ]], dtype=float32),
array([[ 0.06339765, 0.44967934, -0.24905416, -0.86862624],
[ 0.6769087 , -0.22752753, 0.58845747, 0.51635844],
[ 2.7343087 , 2.6904383 , 0.2945323 , 1.9013431 ]],
dtype=float32))
2.2 Build a tiny network#
Now we will start to build a network has one hidden layer of 32 units with ReLU activation.
Output is a single number (regression).
This is equivalent to what we did before with:
MLPRegressor(hidden_layer_sizes=(32,), activation=”relu”)
But the difference is that in scikit-learn the whole pipeline is hidden inside one class.
In PyTorch we build the pieces ourselves: the layers, the activation, and the forward pass.
This way you can see what is happening under the hood.
How PyTorch models work
A model is a Python object with layers. Each layer has parameters (weights and bias).
You pass an input tensor through the model to get predictions. This call is the forward pass.
PyTorch records operations during the forward pass. That record lets it compute gradients during
loss.backward().Parameters live in
model.parameters()and can be saved withmodel.state_dict()..to(device)moves the model to GPU if available. Inputs must be on the same device.
Two common ways to build a model
nn.Sequential: fast for simple stacks.Subclass
nn.Module: gives you a customforwardmethod and more control.
Below we show both styles. Pick one. They behave the same here.
# Sequential style: quickest for simple feed-forward nets
in_dim = X_tr_s.shape[1]
reg_model = nn.Sequential(
nn.Linear(in_dim, 32), # weights W: [in_dim, 32], bias b: [32]
nn.ReLU(),
nn.Linear(32, 1) # weights W: [32, 1], bias b: [1]
)
reg_model
Sequential(
(0): Linear(in_features=4, out_features=32, bias=True)
(1): ReLU()
(2): Linear(in_features=32, out_features=1, bias=True)
)
# Inspect shapes of parameters
for name, p in reg_model.named_parameters():
print(f"{name:20s} shape={tuple(p.shape)} requires_grad={p.requires_grad}")
0.weight shape=(32, 4) requires_grad=True
0.bias shape=(32,) requires_grad=True
2.weight shape=(1, 32) requires_grad=True
2.bias shape=(1,) requires_grad=True
A custom module version looks like this:
class TinyRegressor(nn.Module):
def __init__(self, in_dim, hidden=32):
super().__init__()
self.fc1 = nn.Linear(in_dim, hidden) # input -> hidden layer
self.act = nn.ReLU() # ReLU activation function in hidden layer
self.fc2 = nn.Linear(hidden, 1) # hidden -> output layer
def forward(self, x):
# x has shape [batch, in_dim]
h = self.act(self.fc1(x)) # shape [batch, hidden]
out = self.fc2(h) # shape [batch, 1]
return out
reg_model2 = TinyRegressor(in_dim, hidden=32)
reg_model2
TinyRegressor(
(fc1): Linear(in_features=4, out_features=32, bias=True)
(act): ReLU()
(fc2): Linear(in_features=32, out_features=1, bias=True)
)
Shapes to keep in mind
Input batch
x:[batch, in_dim]Hidden layer output:
[batch, 32]Final output:
[batch, 1]
Tip: you can print a few predictions to sanity check the flow (numbers will be random before training).
x_sample = torch.from_numpy(X_tr_s[:3])
with torch.no_grad():
print("Raw outputs using first 3 values on X-train:", reg_model(x_sample).cpu().numpy().ravel())
Raw outputs using first 3 values on X-train: [-0.3615578 0.06239668 0.7558894 ]
For the rest of this chapter, we will use method 2 (reg_model2) to show the rest steps.
2.3 Loss and optimizer#
For regression we use MSELoss.
The optimizer is Adam, which updates weights smoothly.
loss_fn = nn.MSELoss()
optimizer = torch.optim.Adam(reg_model2.parameters(), lr=1e-2, weight_decay=1e-3)
optimizer
Adam (
Parameter Group 0
amsgrad: False
betas: (0.9, 0.999)
capturable: False
differentiable: False
eps: 1e-08
foreach: None
fused: None
lr: 0.01
maximize: False
weight_decay: 0.001
)
2.4 One training step demo#
To see the loop clearly: forward → loss → backward → update.
xb = torch.from_numpy(X_tr_s[:64])
yb = torch.from_numpy(y_tr[:64])
pred = reg_model2(xb) # forward pass
loss = loss_fn(pred, yb) # compute loss
optimizer.zero_grad() # clear old grads
loss.backward() # compute new grads for each parameter
optimizer.step() # apply the update
float(loss.item())
19706.015625
2.5 Training loop#
We train for 150 epochs. Each epoch goes through the dataset in batches.
We plot the loss to see if the model is learning.
class NumpyDataset(Dataset):
def __init__(self, X, y):
self.X = torch.from_numpy(X).float()
self.y = torch.from_numpy(y).float()
def __len__(self): return len(self.X)
def __getitem__(self, i): return self.X[i], self.y[i]
train_loader = DataLoader(NumpyDataset(X_tr_s, y_tr), batch_size=64, shuffle=True)
train_losses = []
reg_model2.train()
for epoch in range(150):
batch_losses = []
for xb, yb in train_loader:
xb, yb = xb, yb
pred = reg_model2(xb)
loss = loss_fn(pred, yb)
optimizer.zero_grad(); loss.backward(); optimizer.step()
batch_losses.append(loss.item())
train_losses.append(np.mean(batch_losses))
plt.figure(figsize=(5,3))
plt.plot(train_losses)
plt.xlabel("epoch"); plt.ylabel("train MSE")
plt.title("Training loss - regression")
plt.grid(alpha=0.3)
plt.show()
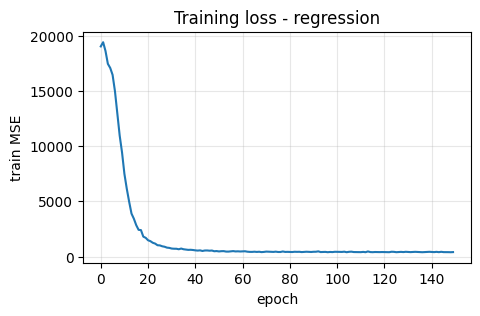
Let’s break down what happened above step by step:
Dataset and DataLoader
We wrapped our numpy arrays inNumpyDataset, which makes them look like a PyTorch dataset.
ThenDataLoadersplit the dataset into mini-batches of 64 rows and shuffled them each epoch.
This helps training be faster and more stable.Epochs and batches
Eachepochmeans “one full pass over the training data”.
Inside each epoch, we looped over mini-batches. For every batch:We ran the forward pass:
pred = reg_model(xb)We computed the loss:
loss = loss_fn(pred, yb)We reset old gradients with
optimizer.zero_grad()We called
loss.backward()so PyTorch computes gradients for each weightWe called
optimizer.step()to update the weights slightly
Loss tracking
We stored the average loss per epoch intrain_losses.
If you plottrain_losses, you should see it go down.
This means the network predictions are getting closer to the true labels.
By the end of 150 epochs, the model should be much better than at the start.
2.6 Evaluate regression#
We check mean squared error and plot predicted vs true Meltig Point.
reg_model2.eval()
with torch.no_grad():
yhat_te = reg_model2(torch.from_numpy(X_te_s)).cpu().numpy()
from sklearn.metrics import mean_squared_error, r2_score
print("MSE:", mean_squared_error(y_te, yhat_te))
print("R2:", r2_score(y_te, yhat_te))
plt.figure(figsize=(4.6,4))
plt.scatter(y_te, yhat_te, alpha=0.6)
lims = [min(y_te.min(), yhat_te.min()), max(y_te.max(), yhat_te.max())]
plt.plot(lims, lims, "k--")
plt.xlabel("True logS"); plt.ylabel("Pred logS")
plt.title("Parity plot")
plt.show()
MSE: 377.2238
R2: 0.8732098882942341
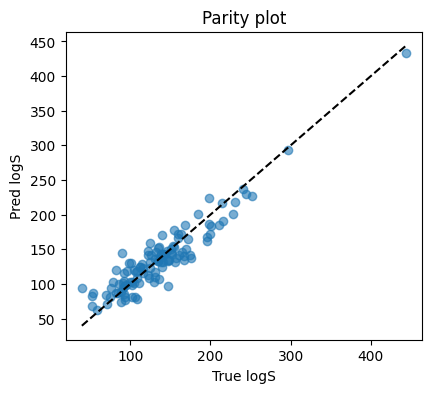
This suggests our customized NN achieve a good performance on this task :D
2.7 Baseline comparison#
To understand how our PyTorch neural network compares to standard regressors, we evaluate three baselines:
Decision Tree Regressor: a single non-linear tree.
Random Forest Regressor: an ensemble of decision trees.
MLPRegressor (sklearn): configured to closely match our PyTorch NN.
Our PyTorch model is:
Input: 4 features
Hidden layer: 32 units with ReLU
Output: 1 unit
Optimizer: Adam with learning rate 1e-2 and weight decay 1e-3
Trained for 150 epochs with mini-batches of size 64.
To mirror this in scikit-learn, we set:
hidden_layer_sizes=(32,): one hidden layer of 32 units, just like PyTorch.activation="relu": same nonlinearity.solver="adam": same optimization family.learning_rate_init=1e-2: match the PyTorch learning rate.alpha=1e-3: equivalent to weight decay 1e-3.max_iter=2000: roughly comparable to 150 epochs in Torch.
We then compare all four models (Decision Tree, Random Forest, sklearn MLP, and PyTorch NN) on the test set using MSE and R².
⏰ Exercises 2.x
For our own NN by Pytorch change the hidden size to 64 and rerun. Does R² improve.
Increase
weight_decayto1e-2. What happens to train vs test scores.
You need to re-initialize the optimizer and re-run the training loop, as there is no
.fit(...)function for our customized NN. In next section you will see we can wrap this logic into a helper function to avoid repetition.
2.8 PyTorch Regressor with sklearn-like Interface#
In earlier sections, we trained our custom PyTorch model by manually writing the training loop. How can we make our customized NN feel more convenient?
We can introduce a small wrapper called CHEM5080Regressor that makes our
PyTorch model behave like a sklearn regressor. The class builds a Sequential
network with two hidden layers (64 units with ReLU, then 32 units with Tanh),
and provides:
.fit(X, y)for training.predict(X)for inference
This way, the PyTorch model can be plugged into the same workflow as Decision
Tree, Random Forest, or sklearn’s MLP. You can customize the architecture inside
nn.Sequential by changing the hidden sizes or swapping the activation
functions.
class NumpyDataset(Dataset):
def __init__(self, X, y):
self.X = torch.from_numpy(X).float()
self.y = torch.from_numpy(y).float()
def __len__(self): return len(self.X)
def __getitem__(self, i): return self.X[i], self.y[i]
class CHEM5080Regressor:
"""
A simple sklearn-like wrapper around a PyTorch feed-forward regressor.
Architecture:
Input -> Linear(4, 64) -> ReLU
-> Linear(64, 32) -> ReLU
-> Linear(32, 1)
"""
def __init__(self, in_dim, lr=1e-2, weight_decay=1e-3, epochs=150, batch_size=64):
# Build Sequential model
self.model = nn.Sequential(
nn.Linear(in_dim, 64),
nn.ReLU(), # Hidden layer 1
nn.Linear(64, 32),
nn.ReLU(), # Hidden layer 2
nn.Linear(32, 1) # Output
)
# Training settings
self.lr = lr
self.weight_decay = weight_decay
self.epochs = epochs
self.batch_size = batch_size
self.loss_fn = nn.MSELoss()
self.optimizer = torch.optim.Adam(self.model.parameters(), lr=lr, weight_decay=weight_decay)
def fit(self, X, y):
dataset = NumpyDataset(X, y)
loader = DataLoader(dataset, batch_size=self.batch_size, shuffle=True)
self.model.train()
for epoch in range(self.epochs):
for xb, yb in loader:
pred = self.model(xb)
loss = self.loss_fn(pred, yb)
self.optimizer.zero_grad()
loss.backward()
self.optimizer.step()
return self
def predict(self, X):
self.model.eval()
with torch.no_grad():
X_tensor = torch.from_numpy(X).float()
return self.model(X_tensor).cpu().numpy().ravel()
# --- Train and evaluate ---
chem_model = CHEM5080Regressor(in_dim=X_tr_s.shape[1], epochs=150)
chem_model.fit(X_tr_s, y_tr)
y_pred = chem_model.predict(X_te_s)
print("MSE:", mean_squared_error(y_te, y_pred))
print("R2:", r2_score(y_te, y_pred))
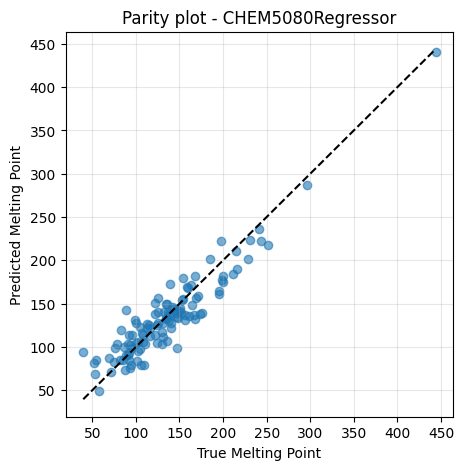
MSE: 387.48224
R2: 0.8697618698517982
It works exactly the same way as we see in previous lectures with sklearn, but it was written by ourselves!
Show code cell source
# --- Optional: Parity plot ---
plt.figure(figsize=(5,5))
plt.scatter(y_te, y_pred, alpha=0.6)
lims = [min(y_te.min(), y_pred.min()), max(y_te.max(), y_pred.max())]
plt.plot(lims, lims, "k--")
plt.xlabel("True Melting Point")
plt.ylabel("Predicted Melting Point")
plt.title("Parity plot - CHEM5080Regressor")
plt.grid(alpha=0.3)
plt.show()
3. Molecules as graphs#
3.1 Why Graph?#
Graphs are sets of vertices connected by edges.
Graphs are general language/data structure for describing and analyzing relational/interacting objects.
Graph machine learning tasks:
Graph-level prediction (Our focus today)
Node-level prediction
Edge-level prediction
Generation
Subgraph-level tasks
Now let’s look at a few examples. We first define a helper function called draw_graph_structure() which will take adjacency matrix to construct a graph.
Show code cell source
# Get and helper function and routine to draw graph structure
def draw_graph_structure(adjacency_matrix, color=None, edge_attr=None):
G = nx.Graph()
n_node = adjacency_matrix.shape[0]
for i in range(n_node):
for j in range(i):
if adjacency_matrix[i, j]:
if edge_attr is not None:
G.add_edge(i, j, weight=edge_attr[i, j])
else:
G.add_edge(i, j)
pos = nx.spring_layout(G, seed=0)
plt.figure(figsize=(5,5)) # keep fixed 5x5 size
# Draw nodes
if color is None:
nx.draw_networkx_nodes(G, pos, node_color="lightblue")
else:
nx.draw_networkx_nodes(G, pos, node_color=color.flatten(), cmap=plt.cm.Set1)
# Draw edges
if edge_attr is not None:
weights = [G[u][v]['weight'] for u,v in G.edges()]
nx.draw_networkx_edges(G, pos, edge_color=weights, edge_cmap=plt.cm.Blues, width=2)
# Optionally add edge labels
edge_labels = {(u, v): f"{G[u][v]['weight']:.1f}" for u, v in G.edges()}
nx.draw_networkx_edge_labels(G, pos, edge_labels=edge_labels, font_size=8)
else:
nx.draw_networkx_edges(G, pos)
nx.draw_networkx_labels(G, pos)
plt.axis("off")
plt.show()
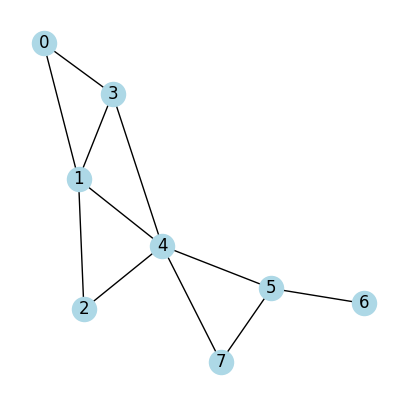
Case 1:
All nodes are identical
All edges are identical
# An adjacency matrix define a graph
# A (adjacency matrix) of shape (num_nodes, num_nodes)
A = np.array([[0,1,0,1,0,0,0,0], #Node 0 connects to Node 1 (second) and Node 3 (fourth)
[1,0,1,1,1,0,0,0], #Node 1 connects to Node 0 (first) and Node 2,3,4 (third to sixth on list)
[0,1,0,0,1,0,0,0], #...
[1,1,0,0,1,0,0,0],
[0,1,1,1,0,1,0,1],
[0,0,0,0,1,0,1,1],
[0,0,0,0,0,1,0,0],
[0,0,0,0,1,1,0,0]]);
print(A)
draw_graph_structure(A)
[[0 1 0 1 0 0 0 0]
[1 0 1 1 1 0 0 0]
[0 1 0 0 1 0 0 0]
[1 1 0 0 1 0 0 0]
[0 1 1 1 0 1 0 1]
[0 0 0 0 1 0 1 1]
[0 0 0 0 0 1 0 0]
[0 0 0 0 1 1 0 0]]
Case 2:
Nodes are not identical
All edges are identical
# Node features and adjacency matrix define a graph
# A (adjacency matrix) of shape (num_nodes, num_nodes)
# H (Node features matrix) of shape (num_nodes, dim_nodes)
A = np.array([[0,1,0,1,0,0,0,0],
[1,0,1,1,1,0,0,0],
[0,1,0,0,1,0,0,0],
[1,1,0,0,1,0,0,0],
[0,1,1,1,0,1,0,1],
[0,0,0,0,1,0,1,1],
[0,0,0,0,0,1,0,0],
[0,0,0,0,1,1,0,0]])
H = np.array([
[1],
[0],
[1],
[2],
[2],
[0],
[1],
[1]
])
print(H)
print(A)
draw_graph_structure(A, color=H)
[[1]
[0]
[1]
[2]
[2]
[0]
[1]
[1]]
[[0 1 0 1 0 0 0 0]
[1 0 1 1 1 0 0 0]
[0 1 0 0 1 0 0 0]
[1 1 0 0 1 0 0 0]
[0 1 1 1 0 1 0 1]
[0 0 0 0 1 0 1 1]
[0 0 0 0 0 1 0 0]
[0 0 0 0 1 1 0 0]]
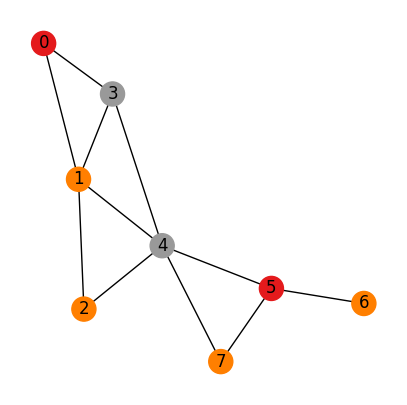
Case 3:
Nodes are not identical
Edges are not identical
# An adjacency matrix define a graph
# A (adjacency matrix) of shape (num_nodes, num_nodes)
# H (Node features matrix) of shape (num_nodes, dim_nodes)
# E (Edge features tensor) of shape (num_nodes, num_nodes, dim_edges)
A = np.array([[0,1,0,1,0,0,0,0],
[1,0,1,1,1,0,0,0],
[0,1,0,0,1,0,0,0],
[1,1,0,0,1,0,0,0],
[0,1,1,1,0,1,0,1],
[0,0,0,0,1,0,1,1],
[0,0,0,0,0,1,0,0],
[0,0,0,0,1,1,0,0]])
H = np.array([
[1],
[0],
[1],
[2],
[2],
[0],
[1],
[1]
])
E = np.zeros((H.shape[0], H.shape[0], 2))
for i in range(H.shape[0]):
for j in range(H.shape[0]):
if A[i, j] == 1:
E[i, j, 0] = H[i, 0]
E[i, j, 1] = H[j, 0]
print(f"Adjacency matrix: \n{A}")
#print(f"Node features matrix: \n{H}")
#print(f"Edge features tensor: \n{E}")
draw_graph_structure(A, color=H, edge_attr=E[:,:,0])
Adjacency matrix:
[[0 1 0 1 0 0 0 0]
[1 0 1 1 1 0 0 0]
[0 1 0 0 1 0 0 0]
[1 1 0 0 1 0 0 0]
[0 1 1 1 0 1 0 1]
[0 0 0 0 1 0 1 1]
[0 0 0 0 0 1 0 0]
[0 0 0 0 1 1 0 0]]
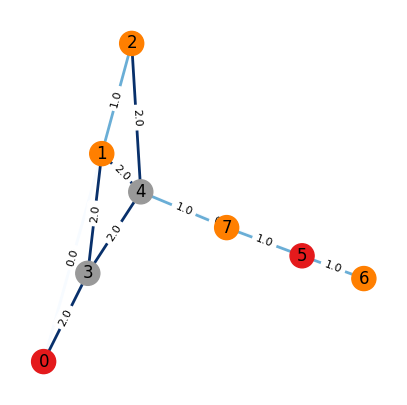
⏰ Exercises 3
Uncomment the two #print statement above the interepret the output information.
3.2 Build tiny toy molecules graphs by hand#
Common node features:
One‑hot element type, degree, formal charge, aromatic flag.
Common edge features:Bond type one‑hot: single, double, triple, aromatic.
We will assemble a small structure that holds:
x: node feature matrix, shape[n_nodes, d_node]edge_index: list of edges as two rows[2, n_edges]edge_attr: edge features[n_edges, d_edge](optional)y: target for the graph
We start with first build a helper function to draw molecule.
Show code cell source
import networkx as nx
def draw_molecule_from_lists(name, atoms, edges_with_bondtypes):
"""
Build x, edge_index, edge_attr from atom + bond lists, draw colored graph,
and print tensor shapes with edge_index table.
"""
# ----- tensors -----
x = np.array(atoms, dtype=np.float32)
edge_index, edge_attr = [], []
for (u, v, b) in edges_with_bondtypes:
edge_index.append((u, v))
edge_index.append((v, u))
edge_attr.append([b])
edge_attr.append([b])
edge_index = np.array(edge_index, dtype=np.int64).T
edge_attr = np.array(edge_attr, dtype=np.int64)
# ----- draw -----
color_map = {
1: "lightgray", # H
6: "lightblue", # C
7: "lightskyblue",# N
8: "lightcoral", # O
9: "lightgreen" # F
}
sym = {1: "H", 6: "C", 7: "N", 8: "O", 9: "F"}
G = nx.Graph()
for i, atom in enumerate(x):
Z = int(atom[0])
G.add_node(
i,
label=f"{sym.get(Z, Z)}{i}",
node_color=color_map.get(Z, "white")
)
for (u, v, b) in edges_with_bondtypes:
G.add_edge(u, v, bond_type=b)
pos = nx.spring_layout(G, seed=17)
labels = nx.get_node_attributes(G, "label")
node_colors = [G.nodes[n]["node_color"] for n in G.nodes()]
nx.draw(G, pos, labels=labels, node_color=node_colors, node_size=1400, font_size=11)
edge_labels = {(u, v): ("double" if d["bond_type"]==2 else "triple" if d["bond_type"]==3 else "single")
for u, v, d in G.edges(data=True)}
nx.draw_networkx_edge_labels(G, pos, edge_labels=edge_labels, font_size=14)
plt.title(name)
plt.axis("off")
plt.show()
# Show tensors as a table
df = pd.DataFrame({
"src": edge_index[0],
"dst": edge_index[1],
"bond_type": edge_attr.flatten()
})
print(f"{name}: x.shape {x.shape}, edge_index.shape {edge_index.shape}, edge_attr.shape {edge_attr.shape}")
print("Helper function [draw_molecule_from_lists] ready to use.")
Helper function [draw_molecule_from_lists] ready to use.
Methane will be our first example.
# ==== Example 1: Methane (CH4) ====
# nodes: 0=C, 1..4=H
atoms_ch4 = [
[6, 4, 0, 0], # C0: atomic number 6, degree 4, not aromatic, neutral
[1, 1, 0, 0], # H1: degree 1
[1, 1, 0, 0], # H2
[1, 1, 0, 0], # H3
[1, 1, 0, 0], # H4
]
edges_ch4 = [
(0, 1, 1), # C0-H1 single
(0, 2, 1), # C0-H2 single
(0, 3, 1), # C0-H3 single
(0, 4, 1), # C0-H4 single
]
draw_molecule_from_lists("Methane (CH4)", atoms_ch4, edges_ch4)
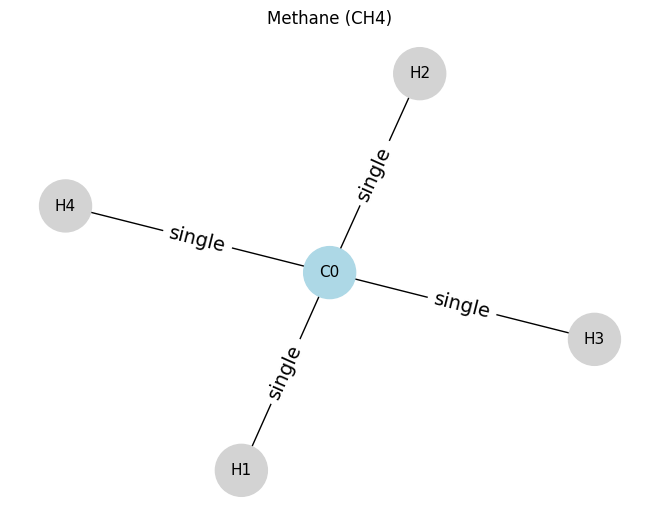
Methane (CH4): x.shape (5, 4), edge_index.shape (2, 8), edge_attr.shape (8, 1)
Then let’s look at ethane. Compare to see the difference.
# ==== Example 2: Ethane (C2H6) ====
# nodes: 0=C0, 1=C1, 2..7=H
atoms_c2h6 = [
[6, 4, 0, 0], # C0
[6, 4, 0, 0], # C1
[1, 1, 0, 0], # H2 on C0
[1, 1, 0, 0], # H3 on C0
[1, 1, 0, 0], # H4 on C0
[1, 1, 0, 0], # H5 on C1
[1, 1, 0, 0], # H6 on C1
[1, 1, 0, 0], # H7 on C1
]
edges_c2h6 = [
(0, 1, 1), # C0-C1 single
(0, 2, 1), # C0-H2 single
(0, 3, 1), # C0-H3 single
(0, 4, 1), # C0-H4 single
(1, 5, 1), # C1-H5 single
(1, 6, 1), # C1-H6 single
(1, 7, 1), # C1-H7 single
]
draw_molecule_from_lists("Ethane (C2H6)", atoms_c2h6, edges_c2h6)
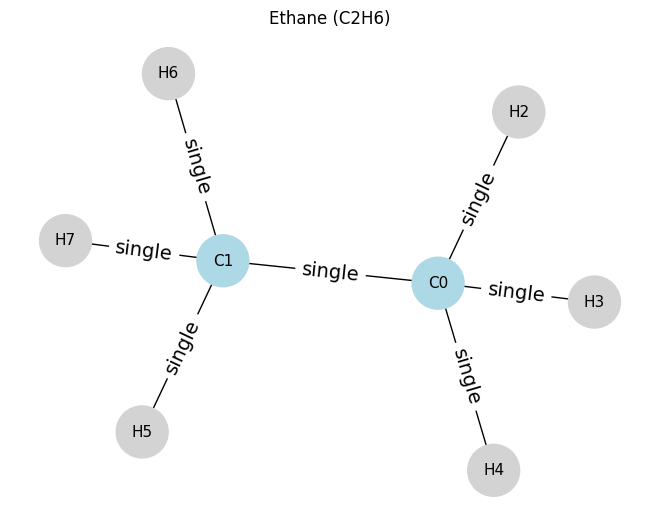
Ethane (C2H6): x.shape (8, 4), edge_index.shape (2, 14), edge_attr.shape (14, 1)
In the example below, we can see it’s also possible to have a different atom and and different bond type.
# ==== Example 3: Formic acid (HCOOH) ====
# nodes: 0=C, 1=O(double), 2=O(single with H3), 3=H on O2, 4=H on C
atoms_hcooh = [
[6, 3, 0, 0], # C0
[8, 1, 0, 0], # O1 (double bond to C0)
[8, 2, 0, 0], # O2 (bonded to C0 and H3)
[1, 1, 0, 0], # H3 (on O2)
[1, 1, 0, 0], # H4 (on C0)
]
edges_hcooh = [
(0, 1, 2), # C0=O1
(0, 2, 1), # C0-O2
(2, 3, 1), # O2-H3
(0, 4, 1), # C0-H4
]
draw_molecule_from_lists("Formic acid (HCOOH)", atoms_hcooh, edges_hcooh)
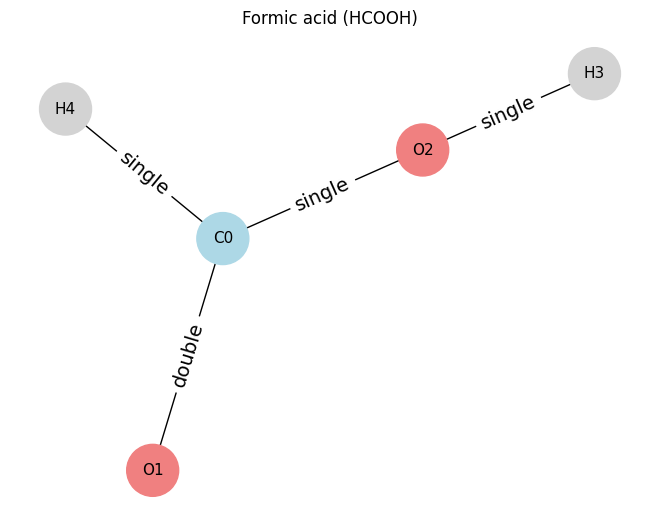
Formic acid (HCOOH): x.shape (5, 4), edge_index.shape (2, 8), edge_attr.shape (8, 1)
# ==== Example 4: Trifluoropropyne (C3F3H) ====
atoms_c3hf3 = [
[6, 4, 0, 0], # C0 (CF3 group carbon)
[6, 2, 0, 0], # C1 (central carbon, part of triple bond)
[6, 1, 0, 0], # C2 (terminal carbon, part of triple bond)
[9, 1, 0, 0], # F3 (on C0)
[9, 1, 0, 0], # F4 (on C0)
[9, 1, 0, 0], # F5 (on C0)
[1, 1, 0, 0], # H6 (on C2)
]
edges_c3hf3 = [
(0,1,1), # C0–C1 single
(1,2,3), # C1≡C2 triple
(0,3,1), # C0–F3 single
(0,4,1), # C0–F4 single
(0,5,1), # C0–F5 single
(2,6,1), # C2–H6 single
]
draw_molecule_from_lists("Trifluoropropyne (CF3–C≡C–H)", atoms_c3hf3, edges_c3hf3)
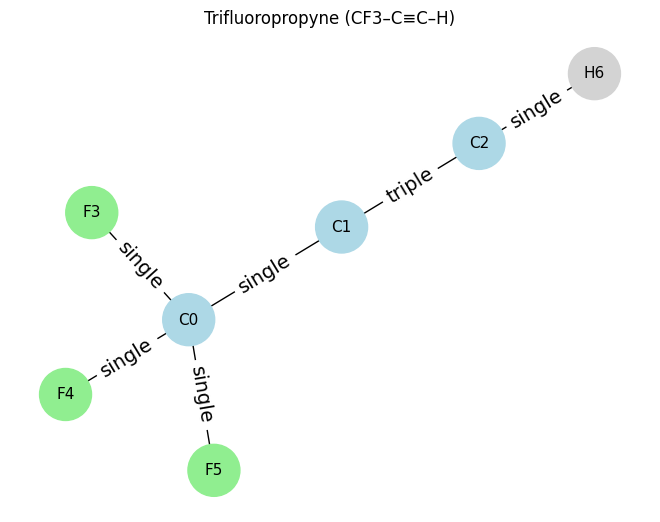
Trifluoropropyne (CF3–C≡C–H): x.shape (7, 4), edge_index.shape (2, 12), edge_attr.shape (12, 1)
⏰ Exercises 3.2
Try to build a graph for Acetonitrile whose smiles string is CC#N (don’t forget about hydrogen atoms!)
3.1 Molecules as graphs#
A molecule can be seen as a graph:
Nodes are atoms with a feature vector per atom.
Edges are bonds that carry types such as single, double, aromatic.
A molecule level label (melting point, toxicity) requires pooling node representations into one vector.
3.1 Minimal node and edge features#
Common node features:
One‑hot element type, degree, formal charge, aromatic flag.
Common edge features:Bond type one‑hot: single, double, triple, aromatic.
We will assemble a small structure that holds:
x: node feature matrix, shape[n_nodes, d_node]edge_index: list of edges as two rows[2, n_edges]edge_attr: edge features[n_edges, d_edge](optional)y: target for the graph
3.2 Build tiny toy graphs by hand#
We start with two toy molecules: Methane and Ethane.
def toy_methane():
# C with 4 H; simple graph centered on C
# nodes: 0=C, 1..4=H
x = np.array([
[6, 4, 0, 0], # very small features: [atomic_number, degree, is_aromatic, formal_charge]
[1, 1, 0, 0],
[1, 1, 0, 0],
[1, 1, 0, 0],
[1, 1, 0, 0],
], dtype=np.float32)
edges = []
for h in [1,2,3,4]:
edges += [(0, h), (h, 0)]
edge_index = np.array(edges, dtype=np.int64).T # shape [2, 8]
return {"x": x, "edge_index": edge_index}
def toy_ethane():
# C-C with 3 H on each carbon
x = np.array([
[6, 4, 0, 0], [6, 4, 0, 0], # two carbons
[1, 1, 0, 0], [1, 1, 0, 0], [1, 1, 0, 0], # Hs on C0
[1, 1, 0, 0], [1, 1, 0, 0], [1, 1, 0, 0], # Hs on C1
], dtype=np.float32)
edges = [(0,1),(1,0)]
for h in [2,3,4]: edges += [(0,h),(h,0)]
for h in [5,6,7]: edges += [(1,h),(h,1)]
edge_index = np.array(edges, dtype=np.int64).T
return {"x": x, "edge_index": edge_index}
g1, g2 = toy_methane(), toy_ethane()
[g1["x"].shape, g1["edge_index"].shape, g2["x"].shape, g2["edge_index"].shape]
[(5, 4), (2, 8), (8, 4), (2, 14)]
Peek at arrays to make sure the shapes are what you expect.
print("Methane x:\n", g1["x"])
print("Methane edges (first 6 cols):\n", g1["edge_index"][:, :6])
Methane x:
[[6. 4. 0. 0.]
[1. 1. 0. 0.]
[1. 1. 0. 0.]
[1. 1. 0. 0.]
[1. 1. 0. 0.]]
Methane edges (first 6 cols):
[[0 1 0 2 0 3]
[1 0 2 0 3 0]]
Tip
Large SMILES → RDKit graphs come later. For now the goal is to see the shapes and write a message passing layer on these toy graphs.
⏰ Exercises 3.x
Add a new toy graph for propane with indices [0,1,2] as the carbon chain and correct hydrogens. Return the same keys x and edge_index. Print its shapes.
4. GNN and MPNN#
Let’s take Methanol (CH₃OH) graph as an example and run both a generic GNN formulation and a more specific MPNN with full 4-dimensional features:
\( h_v^{(0)} = [Z, \text{degree}, \text{aromatic}, \text{charge}] \)
C0:
[6, 4, 0, 0]O1:
[8, 2, 0, 0]H2,H3,H4,H5:
[1, 1, 0, 0]
Edges: C0–H2, C0–H3, C0–H4, C0–O1, O1–H5
GNN (generic form)
Each node aggregates features from its neighbors using a simple rule (often mean or sum), followed by a learnable transformation.
Equation (basic neighborhood aggregation):\[h_v^{(l+1)} = \sigma\big(W_{self} h_v^{(l)} + W_{nei} \cdot \text{AGG}_{u \in N(v)} h_u^{(l)} \big)\]Here \(h_v^{(l)}\) is the feature vector of node \(v\) at layer \(l\), \(N(v)\) is the set of neighbors, and \(\sigma\) is a nonlinearity such as ReLU.
In this form, all neighbors are treated the same and edge features are not used.MPNN (a specific type of GNN)
Each edge can carry information. Messages are computed as functions of both neighbor node states and edge features, then aggregated.
Equations (message passing framework):\[m_{u \to v}^{(l)} = M_\theta(h_u^{(l)}, e_{uv})\]\[h_v^{(l+1)} = U_\theta\Big(h_v^{(l)}, \sum_{u \in N(v)} m_{u \to v}^{(l)}\Big)\]Here \(e_{uv}\) is the feature of edge \((u,v)\) (such as bond type or bond length).
\(M_\theta\) and \(U_\theta\) are neural networks with learnable weights.
Key point
MPNN is not separate from GNN — it is a more specific and chemistry-friendly formulation.
In the generic GNN, neighbors are aggregated without considering edge information.
In MPNN, messages explicitly depend on both neighbor features and edge features, which makes it well suited for molecular graphs.
atoms_ch3oh = [
[6, 4, 0, 0], # C0
[8, 2, 0, 0], # O1
[1, 1, 0, 0], # H2
[1, 1, 0, 0], # H3
[1, 1, 0, 0], # H4
[1, 1, 0, 0], # H5
]
edges_ch3oh = [
(0,1,1), (1,0,1), # C0-O1
(0,2,1), (2,0,1), # C0-H2
(0,3,1), (3,0,1), # C0-H3
(0,4,1), (4,0,1), # C0-H4
(1,5,1), (5,1,1), # O1-H5
]
print("Methanol atoms (node features):", atoms_ch3oh)
print("Methanol edges:", edges_ch3oh)
Methanol atoms (node features): [[6, 4, 0, 0], [8, 2, 0, 0], [1, 1, 0, 0], [1, 1, 0, 0], [1, 1, 0, 0], [1, 1, 0, 0]]
Methanol edges: [(0, 1, 1), (1, 0, 1), (0, 2, 1), (2, 0, 1), (0, 3, 1), (3, 0, 1), (0, 4, 1), (4, 0, 1), (1, 5, 1), (5, 1, 1)]
Note that, different from the previous section, here edges are usually treated as directed, even though chemical bonds are undirected.
Take this methanol bond C0–O1 as an example:
In math, the bond is undirected: {C0, O1}.
But in message passing, information flows in both directions:
C0 → O1 (carbon sends message to oxygen)
O1 → C0 (oxygen sends message to carbon)
So in the edge list (edge_index) we store both directions:
(0,1,1), # C0 → O1, single bond
(1,0,1), # O1 → C0, single bond
The last number 1 is the bond type (single).
One-layer generic (and simple) GNN calculation
Using neighbor mean aggregation:
Example for Carbon (C0):
Neighbors: O1, H2, H3, H4
Mean = \(([8,2,0,0] + [1,1,0,0] + [1,1,0,0] + [1,1,0,0]) / 4 = [2.75, 1.25, 0, 0]\)
Update: \([6,4,0,0] + 0.5\cdot[2.75,1.25,0,0] = [7.375, 4.625, 0, 0]\)
We can compute similarly for O1 and hydrogens.
One-layer MPNN calculation
Messages are weighted by bond strength (inverse bond length, approximate values):
C–H: 0.92
C–O: 0.70
O–H: 1.04
Message function:
Update:
Example for Carbon (C0):
H2 → C0: \([1,1,0,0] \times 0.92 = [0.92,0.92,0,0]\)
H3,H4 similar
O1 → C0: \([8,2,0,0] \times 0.70 = [5.6,1.4,0,0]\)
Sum = [8.36, 4.16, 0, 0]
Update = [6,4,0,0] + 0.5·[8.36,4.16,0,0] = [10.18, 6.08, 0, 0]
When we apply a second layer, information propagates further:
In GNN, all neighbors remain equally weighted.
In MPNN, bond weights amplify or dampen propagation differently (O–H contributes more strongly).
Results after 2 layers will end up with something like this:
GNN: C0 ≈ [10.34, 6.16, 0, 0], O1 ≈ [12.84, 4.91, 0, 0]
MPNN: C0 ≈ [19.09, 11.37, 0, 0], O1 ≈ [16.87, 7.11, 0, 0]
After N layers we compute a pooled graph vector:
This graph vector goes into a final MLP head to predict a property (toxicity, solubility, etc.).
Training adjusts the weights in the update rules and output MLP to minimize a loss function.
We can draw a few molecules graph with features shown on each node before and after updates.
This lets us literally see numbers flowing through edges using below UI.
Note
For web users, please open Colab (link on the top of the page) to run below module. HTML will not be able to update the calculation.
Show code cell source
# Node features = [type_Z, degree, formal_charge, aromatic_flag]
# Edge weights = bond length in Angstroms (Å) [default], with optional inverse-length toggle
# Molecules: Chain, Methane (CH4), Propene (C3H6), Formic acid (HCOOH)
# --------------------------- Utilities ---------------------------
def node_feature_matrix(Z_list, bonds, formal_charges=None, aromatic_flags=None):
"""
Build x from chemistry-style features:
x[i] = [type_Z, degree, formal_charge, aromatic_flag]
- type_Z: atomic number (float)
- degree: number of incident bonds
- formal_charge: default 0
- aromatic_flag: 0/1, default 0
"""
N = len(Z_list)
deg = np.zeros(N, dtype=float)
for u, v, *_ in bonds:
deg[u] += 1.0
deg[v] += 1.0
if formal_charges is None:
formal_charges = np.zeros(N, dtype=float)
if aromatic_flags is None:
aromatic_flags = np.zeros(N, dtype=float)
x = np.stack([np.array(Z_list, dtype=float),
deg,
np.array(formal_charges, dtype=float),
np.array(aromatic_flags, dtype=float)], axis=1)
return x
def expand_edges(bonds):
"""
bonds: list of (u, v, length_A, optional_label)
Return directed edge_index [2,E] and edge_weight [E] (length in Å)
"""
ei = []
ew = []
for b in bonds:
if len(b) == 3:
u, v, L = b
else:
u, v, L, _ = b
ei += [(u, v), (v, u)]
ew += [L, L]
return np.array(ei, dtype=int).T, np.array(ew, dtype=float)
# ----------------------- Molecule builders -----------------------
def mol_methane():
# Nodes: 0=C, 1..4=H; approx C-H 1.09 Å
Z = [6, 1, 1, 1, 1]
bonds = [(0, 1, 1.09), (0, 2, 1.09), (0, 3, 1.09), (0, 4, 1.09)]
x = node_feature_matrix(Z, bonds)
edge_index, edge_w = expand_edges(bonds)
labels = ["C0", "H1", "H2", "H3", "H4"]
colors = ["lightblue", "lightgray", "lightgray", "lightgray", "lightgray"]
return x, edge_index, edge_w, labels, colors
def mol_propene():
# Propene CH3-CH=CH2
# Typical lengths: C–C 1.54 Å, C=C 1.34 Å, C–H 1.09 Å
Z = [6, 6, 6, 1, 1, 1, 1, 1, 1]
bonds = [
(0, 1, 1.54, "C-C"),
(1, 2, 1.34, "C=C"),
(0, 3, 1.09), (0, 4, 1.09), (0, 5, 1.09),
(1, 6, 1.09),
(2, 7, 1.09), (2, 8, 1.09),
]
x = node_feature_matrix(Z, bonds)
edge_index, edge_w = expand_edges(bonds)
labels = ["C0", "C1", "C2", "H3", "H4", "H5", "H6", "H7", "H8"]
colors = ["lightblue"]*3 + ["lightgray"]*6
return x, edge_index, edge_w, labels, colors
def mol_formic_acid():
# Formic acid HCOOH (indices: C0, O1, O2, H3(on O2), H4(on C))
# Typical lengths (Å): C=O 1.23, C–O 1.36, O–H 0.96, C–H 1.09
Z = [6, 8, 8, 1, 1]
bonds = [
(0, 1, 1.23, "C=O"),
(0, 2, 1.36, "C–O"),
(2, 3, 0.96, "O–H"),
(0, 4, 1.09, "C–H"),
]
x = node_feature_matrix(Z, bonds)
edge_index, edge_w = expand_edges(bonds)
labels = ["C0", "O1", "O2", "H3", "H4"]
colors = ["lightblue", "lightcoral", "lightcoral", "lightgray", "lightgray"]
return x, edge_index, edge_w, labels, colors
def mol_methanol():
# Methanol CH3OH
# Typical lengths (Angstrom): C-O 1.43, O-H 0.96, C-H 1.09
# Indices: C0, O1, H2/H3/H4 on C, H5 on O
Z = [6, 8, 1, 1, 1, 1]
bonds = [
(0, 1, 1.43, "C-O"),
(1, 5, 0.96, "O-H"),
(0, 2, 1.09, "C-H"),
(0, 3, 1.09, "C-H"),
(0, 4, 1.09, "C-H"),
]
x = node_feature_matrix(Z, bonds)
edge_index, edge_w = expand_edges(bonds)
labels = ["C0", "O1", "H2", "H3", "H4", "H5"]
colors = ["lightblue", "lightcoral", "lightgray", "lightgray", "lightgray", "lightgray"]
return x, edge_index, edge_w, labels, colors
def graph_chain(): # keep a neutral toy
Z = [0, 0, 0, 0] # type not meaningful here
bonds = [(0,1,1.0),(1,2,1.0),(2,3,1.0)]
x = node_feature_matrix(Z, bonds)
ei = []
for u, v, L in bonds:
ei += [(u,v),(v,u)]
edge_index = np.array(ei, dtype=int).T
edge_w = np.ones(edge_index.shape[1], dtype=float)
labels = [f"Node {i}" for i in range(4)]
colors = None
return x, edge_index, edge_w, labels, colors
BUILDERS = {
"Methane CH4": mol_methane,
"Propene C3H6": mol_propene,
"Formic acid HCOOH": mol_formic_acid,
"Methanol CH3OH": mol_methanol, # <— new
"Chain 0-1-2-3": graph_chain,
}
# -------------------- Message passing math --------------------
def aggregate(x, edge_index, edge_w, how="Mean", weight_mode="Length"):
"""
Aggregate neighbor vectors with edge weights.
- how: "Mean" or "Sum"
- weight_mode: "Length" (multiply by L) or "Inverse length" (multiply by 1/L)
"""
src, dst = edge_index
N, D = x.shape
agg = np.zeros((N, D), dtype=float)
for e in range(src.shape[0]):
w = edge_w[e]
if weight_mode == "Inverse length":
w = 1.0 / max(w, 1e-6)
agg[dst[e]] += w * x[src[e]]
if how == "Mean":
deg = np.zeros(N, dtype=float)
for d in dst:
deg[d] += 1.0
deg = np.maximum(deg, 1.0)
agg = agg / deg[:, None]
return agg
def activate(z, kind="tanh"):
if kind == "Identity": return z
if kind == "ReLU": return np.maximum(z, 0.0)
if kind == "tanh": return np.tanh(z)
return z
def one_step(x, edge_index, edge_w, a=0.6, b=0.8, agg="Mean", act="tanh", safety=True, weight_mode="Length"):
nei = aggregate(x, edge_index, edge_w, how=agg, weight_mode=weight_mode)
z = a * x + b * nei
h = activate(z, kind=act)
if safety:
norms = np.linalg.norm(h, axis=1, keepdims=True) + 1e-8
scale = np.minimum(1.0, 2.0 / norms)
h = h * scale
return h
# ------------------------- Drawing -------------------------
COLORS = ["tab:blue", "tab:orange", "tab:green", "tab:red"]
def show_vectors(vectors, labels, title):
N, D = vectors.shape
fig, ax = plt.subplots(figsize=(1.7*N + 2.0, 3.8))
ax.set_xlim(0, N)
ax.set_ylim(-0.5, 4.7)
ax.axis("off")
ax.set_title(title, pad=10)
for i in range(N):
x_left, x_right = i + 0.1, i + 0.9
ax.plot([x_left, x_left], [0, 4], color="black", linewidth=2)
ax.plot([x_right, x_right], [0, 4], color="black", linewidth=2)
ax.plot([x_left, x_right], [4, 4], color="black", linewidth=2)
ax.plot([x_left, x_right], [0, 0], color="black", linewidth=2)
ax.text((x_left+x_right)/2, 4.3, labels[i], ha="center", va="bottom", fontsize=11)
for k in range(4):
ax.text((x_left+x_right)/2, 3.5 - k, f"[ {vectors[i,k]:+.2f} ]",
ha="center", va="center", fontsize=12, color=COLORS[k])
plt.show()
def show_graph(edge_index, labels, node_colors, title="Molecule/graph connectivity"):
src, dst = edge_index
edges = list(set(tuple(sorted((int(u), int(v)))) for u, v in zip(src, dst) if u != v))
G = nx.Graph()
for i, lab in enumerate(labels):
G.add_node(i, label=lab)
for u, v in edges:
G.add_edge(u, v)
pos = nx.spring_layout(G, seed=7)
fig, ax = plt.subplots(figsize=(5.5, 5.0))
if node_colors is None:
nx.draw(G, pos, with_labels=True, labels={i: labels[i] for i in range(len(labels))},
node_size=1200, font_size=10)
else:
nx.draw(G, pos, with_labels=True, labels={i: labels[i] for i in range(len(labels))},
node_size=1200, font_size=10, node_color=node_colors)
ax.set_title(title)
plt.show()
# ----------------------- Widget UI or Fallback -----------------------
WIDGETS_AVAILABLE = False
try:
import ipywidgets as widgets
from IPython.display import display
WIDGETS_AVAILABLE = True
except Exception:
pass
def run_demo(name="Methane CH4", steps=3, agg="Mean", act="tanh", a=0.6, b=0.8, safety=True, weight_mode="Length"):
x, ei, ew, labels, colors = BUILDERS[name]()
show_graph(ei, labels, colors, title=f"{name} — bonds")
h = x.copy()
for _ in range(steps):
h = one_step(h, ei, ew, a=a, b=b, agg=agg, act=act, safety=safety, weight_mode=weight_mode)
show_vectors(h, labels, title=f"After {steps} step(s) • agg={agg}, act={act}, a={a}, b={b}, safety={safety}, weight={weight_mode}")
if WIDGETS_AVAILABLE:
graph_dd = widgets.Dropdown(options=list(BUILDERS.keys()), value="Methanol CH3OH", description="Molecule")
steps_sl = widgets.IntSlider(min=0, max=10, step=1, value=0, description="Steps")
agg_tb = widgets.ToggleButtons(options=["Mean","Sum"], value="Sum", description="Aggregate")
act_tb = widgets.ToggleButtons(options=["tanh","ReLU","Identity"], value="Identity", description="Activation")
a_sl = widgets.FloatSlider(min=0.0, max=1.5, step=0.05, value=1.0, description="Self (a)")
b_sl = widgets.FloatSlider(min=0.0, max=1.5, step=0.05, value=0.5, description="Neighbor (b)")
safety_cb = widgets.Checkbox(value=False, description="Renormalization / Safety Scale")
weight_tb = widgets.ToggleButtons(options=["Length","Inverse length"], value="Inverse length", description="Edge feature")
run_btn = widgets.Button(description="Run")
out_graph = widgets.Output()
out_vec = widgets.Output()
def on_run(_):
out_graph.clear_output(wait=True); out_vec.clear_output(wait=True)
with out_graph:
x, ei, ew, labels, colors = BUILDERS[graph_dd.value]()
show_graph(ei, labels, colors, title=f"{graph_dd.value} — bonds (Å weighting)")
with out_vec:
h = x.copy()
for _ in range(steps_sl.value):
h = one_step(h, ei, ew, a=a_sl.value, b=b_sl.value,
agg=agg_tb.value, act=act_tb.value,
safety=safety_cb.value, weight_mode=weight_tb.value)
show_vectors(h, labels, title=f"After {steps_sl.value} step(s) • agg={agg_tb.value}, act={act_tb.value}, a={a_sl.value}, b={b_sl.value}, safety={safety_cb.value}, weight={weight_tb.value}")
run_btn.on_click(on_run)
display(widgets.VBox([
widgets.HBox([graph_dd, steps_sl, weight_tb]),
widgets.HBox([agg_tb, act_tb]),
widgets.HBox([a_sl, b_sl, safety_cb, run_btn]),
widgets.HBox([out_graph, out_vec])
]))
on_run(None)
else:
# Fallback run here so you can see the revised initialization and new Formic acid example
run_demo(name="Methanol CH3OH", steps=0, agg="Mean", act="Identity", a=1.0, b=0.5, safety=False, weight_mode="Inverse length")
5. Build an MPNN from scratch#
Now that we’ve seen the math for message passing, we will implement a simple MPNN layer in PyTorch and use it for a toxicity classification task.
At each layer t, every node gets messages from its neighbors and updates its hidden state.
A simple form:
h_vis the node vector at layert.W_selfmaps the node to itself.W_neimaps neighbor messages.Sum neighbor messages, then apply a nonlinearity
σsuch as ReLU.
After T layers, pool all node vectors to get a graph vector:
POOL can be sum, mean, or max. For regression we feed h_graph to a linear head to predict a scalar.
We will use RDKit to parse SMILES strings and turn molecules into graphs.
Show code cell source
def smiles_to_graph(smiles):
mol = Chem.MolFromSmiles(smiles)
if mol is None:
raise ValueError(f"Invalid SMILES: {smiles}")
# Ensure we have atom indices in the depiction later
mol = Chem.AddHs(mol) # to make hydrogens explicit for clearer tables
Chem.SanitizeMol(mol)
x = []
for atom in mol.GetAtoms():
x.append([
atom.GetAtomicNum(),
atom.GetDegree(), # degree counts explicit neighbors
atom.GetFormalCharge(),
1 if atom.GetIsAromatic() else 0
])
edge_index = []
edge_attr = []
bond_type_map = {
Chem.rdchem.BondType.SINGLE: 1,
Chem.rdchem.BondType.DOUBLE: 2,
Chem.rdchem.BondType.TRIPLE: 3,
Chem.rdchem.BondType.AROMATIC: 0
}
for bond in mol.GetBonds():
u, v = bond.GetBeginAtomIdx(), bond.GetEndAtomIdx()
bt = bond_type_map.get(bond.GetBondType(), 0)
edge_index += [(u, v), (v, u)]
edge_attr += [[bt], [bt]]
x = np.array(x, dtype=np.float32)
edge_index = np.array(edge_index, dtype=np.int64).T # [2, E]
edge_attr = np.array(edge_attr, dtype=np.int64) # [E, 1]
return mol, x, edge_index, edge_attr
def atom_df(mol, x):
rows = []
for i, atom in enumerate(mol.GetAtoms()):
rows.append({
"idx": i,
"symbol": atom.GetSymbol(),
"Z": int(x[i,0]),
"degree": int(x[i,1]),
"formal_charge": int(x[i,2]),
"aromatic": bool(x[i,3]),
})
return pd.DataFrame(rows)
def bond_df(mol, edge_index, edge_attr):
# collapse directed edges to undirected unique bonds
seen = set()
rows = []
for b in mol.GetBonds():
u, v = b.GetBeginAtomIdx(), b.GetEndAtomIdx()
key = tuple(sorted((u, v)))
if key in seen:
continue
seen.add(key)
bt = b.GetBondType().name
rows.append({
"u": key[0],
"v": key[1],
"bond_type": bt
})
return pd.DataFrame(rows)
def draw_mol(mol, title):
try:
img = Draw.MolToImage(mol, size=(400, 300), kekulize=True)
plt.figure(figsize=(4,3))
plt.imshow(img)
plt.axis("off")
plt.title(title)
plt.show()
except:
# Fallback: NetworkX from connectivity
import networkx as nx
G = nx.Graph()
for atom in mol.GetAtoms():
G.add_node(atom.GetIdx(), label=f"{atom.GetSymbol()}{atom.GetIdx()}")
for bond in mol.GetBonds():
u, v = bond.GetBeginAtomIdx(), bond.GetEndAtomIdx()
G.add_edge(u, v)
pos = nx.spring_layout(G, seed=7)
labels = nx.get_node_attributes(G, "label")
plt.figure(figsize=(4,3))
nx.draw(G, pos, with_labels=True, labels=labels, node_size=800)
plt.title(title)
plt.show()
Show code cell source
examples = {
"Ethanol (CCO)": "CCO",
"Acetonitrile (CC#N)": "CC#N",
"Formic acid (OC=O)": "OC=O",
"Propene (C=CC)": "C=CC",
}
for name, smi in examples.items():
try:
mol, x, ei, ea = smiles_to_graph(smi)
# DataFrames
adf = atom_df(mol, x)
bdf = bond_df(mol, ei, ea)
# Shapes
print(f'Example: {name}')
print(f"x.shape {x.shape}, edge_index.shape {ei.shape}, edge_attr.shape {ea.shape}")
print("Atom features:", x)
print("Bond features (first three):", edge_attr[:3])
# Drawing (with atom indices rendered by RDKit via AddHs)
draw_mol(mol, f"{name} (with explicit H)")
print("------------------------------------")
except Exception as e:
print(f"{name}: error -> {e}")
Example: Ethanol (CCO)
x.shape (9, 4), edge_index.shape (2, 16), edge_attr.shape (16, 1)
Atom features: [[6. 4. 0. 0.]
[6. 4. 0. 0.]
[8. 2. 0. 0.]
[1. 1. 0. 0.]
[1. 1. 0. 0.]
[1. 1. 0. 0.]
[1. 1. 0. 0.]
[1. 1. 0. 0.]
[1. 1. 0. 0.]]
Ethanol (CCO): error -> name 'edge_attr' is not defined
Example: Acetonitrile (CC#N)
x.shape (6, 4), edge_index.shape (2, 10), edge_attr.shape (10, 1)
Atom features: [[6. 4. 0. 0.]
[6. 2. 0. 0.]
[7. 1. 0. 0.]
[1. 1. 0. 0.]
[1. 1. 0. 0.]
[1. 1. 0. 0.]]
Acetonitrile (CC#N): error -> name 'edge_attr' is not defined
Example: Formic acid (OC=O)
x.shape (5, 4), edge_index.shape (2, 8), edge_attr.shape (8, 1)
Atom features: [[8. 2. 0. 0.]
[6. 3. 0. 0.]
[8. 1. 0. 0.]
[1. 1. 0. 0.]
[1. 1. 0. 0.]]
Formic acid (OC=O): error -> name 'edge_attr' is not defined
Example: Propene (C=CC)
x.shape (9, 4), edge_index.shape (2, 16), edge_attr.shape (16, 1)
Atom features: [[6. 3. 0. 0.]
[6. 3. 0. 0.]
[6. 4. 0. 0.]
[1. 1. 0. 0.]
[1. 1. 0. 0.]
[1. 1. 0. 0.]
[1. 1. 0. 0.]
[1. 1. 0. 0.]
[1. 1. 0. 0.]]
Propene (C=CC): error -> name 'edge_attr' is not defined
Now, let’s define a simple MPNN. Each node aggregates neighbor messages weighted by edge type embeddings.
Below you will see it has two parts:
MPNNLayer
This is a single message-passing step.
Its job is to update each node’s representation by combining:
The node’s own features (W_self(x)).
Messages from its neighbors (W_nei(pre_msg)), where edges add context.
The layer outputs a new embedding for every node in the graph: [N, out_dim].
Think of it like one “round of communication” between nodes.
MPNNClassifier
This is the full model that stacks multiple MPNNLayers to capture more complex dependencies.
self.layer1 → does the first message passing.
self.layer2 → refines node embeddings further.
After the message passing, you now have updated node features (h).
But for graph classification you need a single vector for the whole graph. That’s where pooling comes in:
h_graph = h.mean(dim=0)
This aggregates node embeddings into one graph embedding.
Finally, self.fc maps that graph embedding to class logits (n_classes).
class MPNNLayer(nn.Module):
def __init__(self, in_dim, out_dim, n_edge_types=4):
super().__init__()
self.W_self = nn.Linear(in_dim, out_dim)
self.W_nei = nn.Linear(in_dim, out_dim)
self.edge_emb = nn.Embedding(n_edge_types, in_dim) # embeds to in_dim
def forward(self, x, edge_index, edge_attr):
# x: [N, in_dim], edge_index: [2, E], edge_attr: [E, 1] (long)
row, col = edge_index # messages from row -> col
edge_feat = self.edge_emb(edge_attr.squeeze()) # [E, in_dim]
# message computation (still in in_dim before W_nei)
pre_msg = x[row] + edge_feat # [E, in_dim]
messages = self.W_nei(pre_msg) # [E, out_dim]
# aggregate into [N, out_dim] on the same device and dtype
out_dim = self.W_nei.out_features
agg = x.new_zeros(x.size(0), out_dim) # [N, out_dim]
agg.index_add_(0, col, messages) # sum over incoming edges
out = self.W_self(x) + agg # [N, out_dim]
return torch.relu(out)
class MPNNClassifier(nn.Module):
def __init__(self, in_dim, hidden=64, n_classes=2):
super().__init__()
self.layer1 = MPNNLayer(in_dim, hidden)
self.layer2 = MPNNLayer(hidden, hidden)
self.fc = nn.Linear(hidden, n_classes)
def forward(self, data):
x, edge_index, edge_attr = data["x"], data["edge_index"], data["edge_attr"]
h = self.layer1(x, edge_index, edge_attr)
h = self.layer2(h, edge_index, edge_attr)
h_graph = h.mean(dim=0) # mean pooling
return self.fc(h_graph)
# below is an optional sanity check right after defining the layer:
tmp = MPNNLayer(4, 64)
with torch.no_grad():
x = torch.randn(19, 4)
edge_index = torch.tensor([[0,1,2,3],[1,2,3,4]]) # toy edges
edge_attr = torch.zeros(edge_index.size(1), 1, dtype=torch.long)
y = tmp(x, edge_index, edge_attr)
print(y.shape) # should be [19, 64]
torch.Size([19, 64])
We reuse the oxidation dataset, map SMILES → graphs, and toxicity → label.
url = "https://raw.githubusercontent.com/zzhenglab/ai4chem/main/book/_data/C_H_oxidation_dataset.csv"
df_raw = pd.read_csv(url)
label_map = {"toxic":1, "non_toxic":0}
df_clf = df_raw[df_raw["Toxicity"].str.lower().isin(label_map.keys())]
# Convert (mol, x_np, edge_index_np, edge_attr_np) -> dict of torch tensors
def tuple_to_graphdict(g_tuple):
mol, x_np, ei_np, ea_np = g_tuple
g = {
"x": torch.tensor(x_np, dtype=torch.float),
"edge_index": torch.tensor(ei_np, dtype=torch.long),
"edge_attr": torch.tensor(ea_np, dtype=torch.long), # shape [E,1], ints
# keep mol if you want later:
# "mol": mol
}
return g
graphs = []
labels = []
for smi, lab in zip(df_clf["SMILES"], df_clf["Toxicity"].str.lower().map(label_map)):
t = smiles_to_graph(smi) # still returns the 4-tuple
if t is None:
continue
g = tuple_to_graphdict(t) # convert to dict of tensors
g["y"] = torch.tensor(lab, dtype=torch.long)
graphs.append(g)
labels.append(lab)
print("Total graphs:", len(graphs))
Total graphs: 575
We will keep it very small: 80/20 split, train for a few epochs.
train_idx, test_idx = train_test_split(range(len(graphs)), test_size=0.2, random_state=42, stratify=labels)
train_graphs = [graphs[i] for i in train_idx]
test_graphs = [graphs[i] for i in test_idx]
model = MPNNClassifier(in_dim=4, hidden=64, n_classes=2)
optimizer = torch.optim.Adam(model.parameters(), lr=1e-3, weight_decay=1e-4)
loss_fn = nn.CrossEntropyLoss()
def train_epoch(graphs):
model.train()
total_loss = 0
for g in graphs:
optimizer.zero_grad()
out = model(g).unsqueeze(0)
loss = loss_fn(out, g["y"].unsqueeze(0))
loss.backward()
optimizer.step()
total_loss += loss.item()
return total_loss / len(graphs)
def eval_acc(graphs):
model.eval()
correct = 0
with torch.no_grad():
for g in graphs:
out = model(g).unsqueeze(0)
pred = out.argmax(dim=1)
correct += (pred == g["y"]).sum().item()
return correct / len(graphs)
for epoch in range(10):
loss = train_epoch(train_graphs)
acc = eval_acc(test_graphs)
print(f"Epoch {epoch:02d} | Loss {loss:.3f} | Test acc {acc:.3f}")
Epoch 00 | Loss 0.471 | Test acc 0.826
Epoch 01 | Loss 0.423 | Test acc 0.826
Epoch 02 | Loss 0.414 | Test acc 0.826
Epoch 03 | Loss 0.408 | Test acc 0.826
Epoch 04 | Loss 0.403 | Test acc 0.826
Epoch 05 | Loss 0.398 | Test acc 0.826
Epoch 06 | Loss 0.393 | Test acc 0.826
Epoch 07 | Loss 0.387 | Test acc 0.826
Epoch 08 | Loss 0.383 | Test acc 0.817
Epoch 09 | Loss 0.377 | Test acc 0.809
⏰Exercises
Change epoch from 10 to 40 to see the difference.
This gives an test acc around 0.85, which is quite nice (since we have not done any optimization on
The other thing you may notice is that, since we use molecule as graph, this time we did not provide molecular level descritors like MolWt, LogP, TPSA, #Rings, instead we simply just put some simple atomic level information like atomic number and bond order.
Similar to the case you see earlier today with MLP, we can also have customized .fit() and .predict() function.
Show code cell source
def set_seed(seed=42):
torch.manual_seed(seed)
np.random.seed(seed)
def _to_device_graph(g, device):
out = {k: v.to(device) if isinstance(v, torch.Tensor) else v for k, v in g.items()}
return out
class CHEM5080_MPNNClassifierWrapper:
"""
A lightweight sklearn-style wrapper:
- fit(graphs)
- predict(graphs)
- predict_proba(graphs)
- score(graphs, y_true) -> accuracy
Expects each graph g to include g["y"] as a scalar LongTensor (class id).
"""
def __init__(self,
in_dim: int,
n_classes: int,
hidden: int = 64,
lr: float = 1e-3,
weight_decay: float = 1e-4,
epochs: int = 30,
device: str = None,
print_every: int = 1):
self.in_dim = in_dim
self.n_classes = n_classes
self.hidden = hidden
self.lr = lr
self.weight_decay = weight_decay
self.epochs = epochs
self.print_every = print_every
self.device = device or ("cuda" if torch.cuda.is_available() else "cpu")
self.model = MPNNClassifier(in_dim=in_dim, hidden=hidden, n_classes=n_classes).to(self.device)
self.optimizer = torch.optim.Adam(self.model.parameters(), lr=lr, weight_decay=weight_decay)
self.loss_fn = nn.CrossEntropyLoss()
def fit(self, graphs, val_graphs=None):
"""
Trains for self.epochs over the list of graphs.
If val_graphs is provided, reports validation accuracy each epoch.
"""
self.model.train()
best_state = None
best_val = -math.inf
for epoch in range(1, self.epochs + 1):
total_loss = 0.0
self.model.train()
for g in graphs:
g = _to_device_graph(g, self.device)
self.optimizer.zero_grad()
logits = self.model(g).unsqueeze(0) # [1, C]
loss = self.loss_fn(logits, g["y"].unsqueeze(0).to(self.device))
loss.backward()
self.optimizer.step()
total_loss += float(loss.item())
avg_loss = total_loss / max(1, len(graphs))
msg = f"Epoch {epoch:02d} | train_loss {avg_loss:.4f}"
if val_graphs is not None:
acc = self.score(val_graphs)
msg += f" | val_acc {acc:.4f}"
# keep best
if acc > best_val:
best_val = acc
best_state = copy.deepcopy(self.model.state_dict())
if self.print_every and (epoch % self.print_every == 0):
print(msg)
if best_state is not None:
self.model.load_state_dict(best_state)
return self
@torch.no_grad()
def predict_proba(self, graphs):
"""
Returns numpy array of class probabilities with shape [N, n_classes].
"""
self.model.eval()
probs = []
for g in graphs:
g = _to_device_graph(g, self.device)
logits = self.model(g).unsqueeze(0) # [1, C]
p = F.softmax(logits, dim=1).cpu().numpy() # [1, C]
probs.append(p[0])
return np.vstack(probs)
@torch.no_grad()
def predict(self, graphs):
"""
Returns numpy int array of predicted class ids with shape [N].
"""
prob = self.predict_proba(graphs)
return prob.argmax(axis=1).astype(np.int64)
@torch.no_grad()
def score(self, graphs, y_true=None):
"""
Accuracy. If y_true is None, reads g['y'] for each graph.
"""
self.model.eval()
correct = 0
total = 0
if y_true is None:
y_true = [int(g["y"].item()) for g in graphs]
preds = self.predict(graphs)
return float((preds == np.array(y_true)).sum() / len(preds))
# ===== Tiny hyperparameter search (grid) =====
def grid_search_graphclf(
param_grid: dict,
train_graphs: list,
val_graphs: list,
in_dim: int,
n_classes: int,
seed: int = 42,
):
set_seed(seed)
keys = list(param_grid.keys())
# build all combos
from itertools import product
values = [param_grid[k] for k in keys]
combos = [dict(zip(keys, vals)) for vals in product(*values)]
best_acc = -math.inf
best_model = None
best_params = None
history = []
for i, params in enumerate(combos, 1):
cfg = dict(hidden=64, lr=1e-3, weight_decay=1e-4, epochs=30) # defaults
cfg.update(params)
print(f"[{i}/{len(combos)}] testing {cfg}")
model = CHEM5080_MPNNClassifierWrapper(
in_dim=in_dim,
n_classes=n_classes,
hidden=cfg["hidden"],
lr=cfg["lr"],
weight_decay=cfg["weight_decay"],
epochs=cfg["epochs"],
print_every=0
)
model.fit(train_graphs, val_graphs=val_graphs)
acc = model.score(val_graphs)
history.append((cfg, acc))
print(f" val_acc={acc:.4f}")
if acc > best_acc:
best_acc = acc
best_model = model
best_params = cfg
print(f"\nBest val_acc={best_acc:.4f} with params={best_params}")
return best_model, best_params, history
Below you will see we have this new toy CHEM5080_MPNNClassifierWrapper for us to make prediction just like the way you did for lasso, decision tree and random forest in previous lectures.
train_idx, test_idx = train_test_split(
range(len(graphs)), test_size=0.2, random_state=42, stratify=labels
)
train_graphs = [graphs[i] for i in train_idx]
test_graphs = [graphs[i] for i in test_idx]
# Train a single model
clf = CHEM5080_MPNNClassifierWrapper(in_dim=4, n_classes=2, hidden=64, lr=1e-3, weight_decay=1e-4, epochs=10)
clf.fit(train_graphs, val_graphs=test_graphs)
# Evaluate
print("Test accuracy:", clf.score(test_graphs))
# Predict labels and probabilities
y_hat = clf.predict(test_graphs) # int class ids
p_hat = clf.predict_proba(test_graphs)
Epoch 01 | train_loss 0.4850 | val_acc 0.8261
Epoch 02 | train_loss 0.4325 | val_acc 0.8261
Epoch 03 | train_loss 0.4176 | val_acc 0.8261
Epoch 04 | train_loss 0.4117 | val_acc 0.8261
Epoch 05 | train_loss 0.4078 | val_acc 0.8261
Epoch 06 | train_loss 0.4035 | val_acc 0.8261
Epoch 07 | train_loss 0.3995 | val_acc 0.8261
Epoch 08 | train_loss 0.3972 | val_acc 0.8261
Epoch 09 | train_loss 0.3921 | val_acc 0.8261
Epoch 10 | train_loss 0.3865 | val_acc 0.8261
Test accuracy: 0.8260869565217391
We can also try to tune the hyperparameters.
Since running a full search with GNNs is more time-consuming than the other models we saw earlier (like Decision Trees or scikit-learn MLPs), we will just keep the grid small to give you an idea of the tuning process.
Since we run this on CPU, you may find the training relatively slow compared to even feed-forward models, because each epoch processes graphs one by one and the message passing layers are more complex. In practice, GPUs help but do not fully remove this bottleneck — batching and optimized data loaders become more important.
The code below can be excecuted at google colab.
param_grid = {
"hidden": [8, 32, 128],
"lr": [1e-3, 5e-4],
"weight_decay": [1e-4, 5e-5],
"epochs": [30]
}
best_model, best_params, hist = grid_search_graphclf(
param_grid=param_grid,
train_graphs=train_graphs,
val_graphs=test_graphs, # or a proper validation set if you have one
in_dim=4,
n_classes=2,
seed=42
)
print("Best params:", best_params)
print("Best model test accuracy:", best_model.score(test_graphs))
Sample output:
[1/12] testing {‘hidden’: 8, ‘lr’: 0.001, ‘weight_decay’: 0.0001, ‘epochs’: 30} val_acc=0.8348 [2/12] testing {‘hidden’: 8, ‘lr’: 0.001, ‘weight_decay’: 5e-05, ‘epochs’: 30} val_acc=0.8261 [3/12] testing {‘hidden’: 8, ‘lr’: 0.0005, ‘weight_decay’: 0.0001, ‘epochs’: 30} val_acc=0.8348 [4/12] testing {‘hidden’: 8, ‘lr’: 0.0005, ‘weight_decay’: 5e-05, ‘epochs’: 30} val_acc=0.8261 [5/12] testing {‘hidden’: 32, ‘lr’: 0.001, ‘weight_decay’: 0.0001, ‘epochs’: 30} val_acc=0.8435 …
⏰ Exercises 6
Change the number of MPNN layers in MPNNClassifier(). Run again the training and tetsing to see any difference.
7. Glossary#
- graph neural network#
A neural network that operates on graph-structured data, updating node features by aggregating information from neighbors.
- message passing#
The process in a GNN where node representations are updated by exchanging information with neighbors over edges.
- node feature#
A vector describing atom properties such as atomic number, degree, aromaticity, or formal charge.
- edge feature#
A vector describing bond properties such as single/double/triple/aromatic bond type.
- pooling#
Aggregation of all node embeddings into a single graph-level embedding (e.g., mean, sum, or max).
- MPNN#
Message Passing Neural Network. A GNN variant where messages explicitly depend on both neighbor node features and edge features.
8. Quick reference#
Note
Always scale inputs for descriptor-based MLPs.
Start with one hidden layer (32–64 units) before stacking deeper.
For regression: use
MSELoss, check parity plots.For classification: use
CrossEntropyLoss, check confusion matrix and ROC.In GNNs, each edge should be stored in both directions for message passing.
Pool node embeddings (mean/sum/max) to form a graph vector.
Compare with baselines (logistic regression, decision tree, random forest).
Use cross validation when tuning hidden size, learning rate, and epochs.
MLP (PyTorch):
nn.Linear,nn.ReLU,nn.MSELoss,nn.CrossEntropyLoss,torch.optim.AdamMLP (sklearn):
hidden_layer_sizes,activation,alpha(L2),learning_rate_init,max_iterGNN:
x: node feature matrix[n_nodes, d_node]edge_index: edges[2, n_edges](directed)edge_attr: edge features[n_edges, d_edge]Layers: message passing + pooling + MLP head
Evaluation:
Regression:
MSE,MAE,R²Classification:
Accuracy,Precision,Recall,F1,ROC AUC
9. In-class activities#
Q1. Build an MLP for toxicity classification#
Use input features
[MolWt, LogP, TPSA, NumRings].Build an MLP from scratch in PyTorch with one hidden layer of 32 units and ReLU.
Train for 100 epochs for
toxicityprediction.Report accuracy and compare against a logistic regression baseline.
# TO DO
Q2. Build an MLP for regression on melting point#
Same input features
[MolWt, LogP, TPSA, NumRings].Build an MLP with two hidden layers
(64, 32)in PyTorch.Train with MSE loss for 150 epochs for
melting pointprediction.Compare test R² with Random Forest Regressor baseline.
# TO DO
Q3. GNN-1: Toxicity with a slightly different architecture#
Build a GNN with 3 message-passing layers, hidden size 48, sum pooling, and dropout=0.2.
Train on toxicity graphs from SMILES.
Compare test accuracy to a descriptor MLPClassifier baseline.
# TO DO
Q4. GNN-2: Toxicity with 5-fold cross validation#
Build a second GNN that differs again: 2 layers, hidden 96, max pooling.
Run
KFold()orStratifiedKFold()withn_splits=5on toxicity.Report mean accuracy and mean ROC AUC across folds.
# TO DO
Q5. Reactivity classification (MLP from scratch)#
Target =
Reactivity(yes, this is a property exists in our C-H oxidation dataset that we have never touched before! basically it’s binary success/failure labels on the C-H reaction outcome if electrochemical conditions were applied ) in the dataset. Values are−1or1.Map to 0 or 1 and train a PyTorch MLP on
[MolWt, LogP, TPSA, NumRings].Compare against LogisticRegression and RandomForestClassifier baselines.
Report accuracy and ROC AUC.
# TO DO
10. Solutions#
Solution Q1#
# Q1 solution
set_seed(0)
url = "https://raw.githubusercontent.com/zzhenglab/ai4chem/main/book/_data/C_H_oxidation_dataset.csv"
df_oxidation_raw = pd.read_csv(url)
def calc_descriptors(smiles):
mol = Chem.MolFromSmiles(smiles)
if mol is None:
return pd.Series({
"MolWt": None,
"LogP": None,
"TPSA": None,
"NumRings": None
})
return pd.Series({
"MolWt": Descriptors.MolWt(mol), # molecular weight
"LogP": Crippen.MolLogP(mol), # octanol-water logP
"TPSA": rdMolDescriptors.CalcTPSA(mol), # topological polar surface area
"NumRings": rdMolDescriptors.CalcNumRings(mol) # number of rings
})
# Apply the function to the SMILES column
desc_df = df_oxidation_raw["SMILES"].apply(calc_descriptors)
# Concatenate new descriptor columns to original DataFrame
df_clf = pd.concat([df_oxidation_raw, desc_df], axis=1)
df_reg = df_clf.copy()
# Logistic regression baseline
from sklearn.linear_model import LogisticRegression
X = df_clf[["MolWt","LogP","TPSA","NumRings"]].values
y = df_clf["Toxicity"].str.lower().map({"toxic":1,"non_toxic":0}).values
Xtr,Xte,ytr,yte = train_test_split(X,y,test_size=0.2,random_state=42,stratify=y)
baseline = LogisticRegression(max_iter=1000).fit(Xtr,ytr)
print("Baseline acc:", accuracy_score(yte, baseline.predict(Xte)))
# MLP from scratch
class MLP_Q1(nn.Module):
def __init__(self,in_dim,hidden=32,n_classes=2):
super().__init__()
self.fc1 = nn.Linear(in_dim,hidden)
self.fc2 = nn.Linear(hidden,n_classes)
def forward(self,x):
return self.fc2(F.relu(self.fc1(x)))
model = MLP_Q1(4)
opt = torch.optim.Adam(model.parameters(), lr=1e-2)
loss_fn = nn.CrossEntropyLoss()
for epoch in range(200):
xb = torch.tensor(Xtr, dtype=torch.float32)
yb = torch.tensor(ytr, dtype=torch.long)
opt.zero_grad()
out = model(xb)
loss = loss_fn(out,yb)
loss.backward()
opt.step()
model.eval()
ypred = model(torch.tensor(Xte, dtype=torch.float32)).argmax(1).numpy()
print("MLP acc:", accuracy_score(yte, ypred))
#For this question and the following questions, it is fine if the model performance is worse than the baseline.
Baseline acc: 0.9565217391304348
MLP acc: 0.8347826086956521
Solution Q2#
X = df_reg[["MolWt","LogP","TPSA","NumRings"]].values.astype(np.float32)
y = df_reg["Melting Point"].values.astype(np.float32).reshape(-1,1)
Xtr,Xte,ytr,yte = train_test_split(X,y,test_size=0.2,random_state=0)
# Baseline
rf = RandomForestRegressor(n_estimators=200,random_state=0).fit(Xtr,ytr.ravel())
print("Baseline RF R2:", r2_score(yte,rf.predict(Xte)))
# PyTorch MLP
class MLPreg_Q2(nn.Module):
def __init__(self,in_dim):
super().__init__()
self.net = nn.Sequential(
nn.Linear(in_dim,64), nn.ReLU(),
nn.Linear(64,32), nn.ReLU(),
nn.Linear(32,1)
)
def forward(self,x): return self.net(x)
model = MLPreg_Q2(4)
opt = torch.optim.Adam(model.parameters(), lr=1e-3)
loss_fn = nn.MSELoss()
for epoch in range(200):
xb = torch.tensor(Xtr, dtype=torch.float32)
yb = torch.tensor(ytr, dtype=torch.float32)
opt.zero_grad()
loss = loss_fn(model(xb), yb)
loss.backward(); opt.step()
yhat = model(torch.tensor(Xte, dtype=torch.float32)).detach().numpy()
print("MLP R2:", r2_score(yte,yhat))
Baseline RF R2: 0.7318566637682214
MLP R2: 0.7427394813162206
Solution Q3#
# Build toxicity graphs
label_map = {"toxic":1, "non_toxic":0}
df_tox = df[df["Toxicity"].str.lower().isin(label_map.keys())].copy()
y_bin = df_tox["Toxicity"].str.lower().map(label_map).astype(int).values
graphs_tox = []
for smi, yv in zip(df_tox["SMILES"], y_bin):
mol, x_np, ei_np, ea_np = smiles_to_graph(smi)
g = {
"x": torch.tensor(x_np, dtype=torch.float32),
"edge_index": torch.tensor(ei_np, dtype=torch.long),
"edge_attr": torch.tensor(ea_np, dtype=torch.long),
"y": torch.tensor(yv, dtype=torch.long)
}
graphs_tox.append(g)
train_idx, test_idx = train_test_split(
np.arange(len(graphs_tox)),
test_size=0.2, random_state=42, stratify=y_bin
)
train_graphs = [graphs_tox[i] for i in train_idx]
test_graphs = [graphs_tox[i] for i in test_idx]
# Baseline on descriptors: simple MLPClassifier
from sklearn.pipeline import Pipeline
from sklearn.preprocessing import StandardScaler
from sklearn.neural_network import MLPClassifier
from sklearn.metrics import accuracy_score
X_desc = df_tox[["MolWt","LogP","TPSA","NumRings"]].values
Xtr_d, Xte_d = X_desc[train_idx], X_desc[test_idx]
ytr_d, yte_d = y_bin[train_idx], y_bin[test_idx]
mlp_base = Pipeline([
("scaler", StandardScaler()),
("clf", MLPClassifier(hidden_layer_sizes=(16,),
activation="relu",
learning_rate_init=0.01,
max_iter=2000,
random_state=0))
]).fit(Xtr_d, ytr_d)
base_acc = accuracy_score(yte_d, mlp_base.predict(Xte_d))
# GNN variant: 3 layers, sum pooling, dropout
class MPNNClassifierV3(nn.Module):
def __init__(self, in_dim=4, hidden=48, n_layers=3, n_classes=2, dropout=0.2, pool="sum"):
super().__init__()
self.layers = nn.ModuleList()
dims = [in_dim] + [hidden]*(n_layers-1) + [hidden]
for a, b in zip(dims[:-1], dims[1:]):
self.layers.append(MPNNLayer(a, b))
self.dropout = nn.Dropout(dropout)
self.fc = nn.Linear(hidden, n_classes)
self.pool = pool
def forward(self, g):
x, edge_index, edge_attr = g["x"], g["edge_index"], g["edge_attr"]
h = x
for layer in self.layers:
h = layer(h, edge_index, edge_attr)
h = self.dropout(h)
if self.pool == "sum":
h_graph = h.sum(dim=0)
else:
h_graph = h.mean(dim=0)
return self.fc(h_graph)
def train_epoch(model, graphs, opt, loss_fn):
model.train(); total=0
for g in graphs:
opt.zero_grad()
out = model(g).unsqueeze(0)
loss = loss_fn(out, g["y"].unsqueeze(0))
loss.backward(); opt.step()
total += float(loss.item())
return total/len(graphs)
@torch.no_grad()
def eval_acc(model, graphs):
model.eval(); correct=0
for g in graphs:
pred = model(g).argmax().item()
correct += int(pred == g["y"].item())
return correct/len(graphs)
gnn = MPNNClassifierV3(in_dim=4, hidden=48, n_layers=3, dropout=0.2, pool="sum")
opt = torch.optim.Adam(gnn.parameters(), lr=1e-3, weight_decay=1e-4)
loss_fn = nn.CrossEntropyLoss()
for epoch in range(5): #you should replace with 25. Our class website can't run this long training so I put 5 here.
_ = train_epoch(gnn, train_graphs, opt, loss_fn)
gnn_acc = eval_acc(gnn, test_graphs)
print(f"Descriptor MLP baseline acc: {base_acc:.3f}")
print(f"GNN (3 layers, sum pool, dropout) acc: {gnn_acc:.3f}")
Descriptor MLP baseline acc: 0.939
GNN (3 layers, sum pool, dropout) acc: 0.861
Solution Q4#
from sklearn.model_selection import StratifiedKFold
from sklearn.metrics import roc_auc_score
# Prepare once
y_all = np.array([g["y"].item() for g in graphs_tox])
def fit_eval_fold(train_ids, test_ids):
tr = [graphs_tox[i] for i in train_ids]
te = [graphs_tox[i] for i in test_ids]
# Different architecture from Q3 and class: 2 layers, hidden 96, max pooling
class MPNNClassifierCV(nn.Module):
def __init__(self, in_dim=4, hidden=96, n_classes=2):
super().__init__()
self.l1 = MPNNLayer(in_dim, hidden)
self.l2 = MPNNLayer(hidden, hidden)
self.fc = nn.Linear(hidden, n_classes)
def forward(self, g):
x, ei, ea = g["x"], g["edge_index"], g["edge_attr"]
h = self.l1(x, ei, ea)
h = self.l2(h, ei, ea)
h_graph, _ = torch.max(h, dim=0) # max pooling
return self.fc(h_graph)
model = MPNNClassifierCV()
opt = torch.optim.Adam(model.parameters(), lr=1e-3, weight_decay=1e-4)
loss_fn = nn.CrossEntropyLoss()
# quick train
for epoch in range(5): #again, you should replace with 20
model.train()
for g in tr:
opt.zero_grad()
out = model(g).unsqueeze(0)
loss = loss_fn(out, g["y"].unsqueeze(0))
loss.backward(); opt.step()
# eval acc and AUC
model.eval()
correct=0; probs=[]; ys=[]
with torch.no_grad():
for g in te:
logits = model(g)
p = torch.softmax(logits, dim=0)[1].item()
pred = int(logits.argmax().item())
probs.append(p); ys.append(int(g["y"].item()))
correct += int(pred == ys[-1])
acc = correct/len(te)
auc = roc_auc_score(ys, probs)
return acc, auc
skf = StratifiedKFold(n_splits=5, shuffle=True, random_state=0)
accs, aucs = [], []
for tr_ids, te_ids in skf.split(np.arange(len(graphs_tox)), y_all):
acc, auc = fit_eval_fold(tr_ids, te_ids)
accs.append(acc); aucs.append(auc)
print(f"5-fold mean acc: {np.mean(accs):.3f} ± {np.std(accs):.3f}")
print(f"5-fold mean AUC: {np.mean(aucs):.3f} ± {np.std(aucs):.3f}")
Solution Q5#
# Reactivity classification with an MLP from scratch on descriptors
# Reactivity is −1 or 1. Map to {0,1}.
df_rxn = df[["SMILES","Reactivity","MolWt","LogP","TPSA","NumRings"]].dropna().copy()
df_rxn = df_rxn[df_rxn["Reactivity"].isin([-1, 1])]
y_rxn = (df_rxn["Reactivity"].map({-1:0, 1:1})).astype(int).values
X_rxn = df_rxn[["MolWt","LogP","TPSA","NumRings"]].values.astype(np.float32)
Xtr, Xte, ytr, yte = train_test_split(X_rxn, y_rxn, test_size=0.2, random_state=0, stratify=y_rxn)
# Baselines
from sklearn.linear_model import LogisticRegression
from sklearn.ensemble import RandomForestClassifier
from sklearn.metrics import accuracy_score, roc_auc_score
logit = Pipeline([("scaler", StandardScaler()), ("lr", LogisticRegression(max_iter=2000, random_state=0))]).fit(Xtr, ytr)
rf = RandomForestClassifier(n_estimators=300, min_samples_leaf=3, random_state=0, n_jobs=-1).fit(Xtr, ytr)
base_acc_log = accuracy_score(yte, logit.predict(Xte))
base_auc_log = roc_auc_score(yte, logit.predict_proba(Xte)[:,1])
base_acc_rf = accuracy_score(yte, rf.predict(Xte))
base_auc_rf = roc_auc_score(yte, rf.predict_proba(Xte)[:,1])
# PyTorch MLP from scratch
class MLPReact(nn.Module):
def __init__(self, in_dim=4, hidden=(32,16), n_classes=2):
super().__init__()
self.net = nn.Sequential(
nn.Linear(in_dim, hidden[0]), nn.ReLU(),
nn.Linear(hidden[0], hidden[1]), nn.ReLU(),
nn.Linear(hidden[1], n_classes)
)
def forward(self, x): return self.net(x)
scaler = StandardScaler().fit(Xtr)
Xtr_s = scaler.transform(Xtr).astype(np.float32)
Xte_s = scaler.transform(Xte).astype(np.float32)
torch.manual_seed(0)
mlp = MLPReact(in_dim=4, hidden=(32,16))
opt = torch.optim.Adam(mlp.parameters(), lr=1e-3, weight_decay=1e-4)
loss_fn = nn.CrossEntropyLoss()
xb = torch.tensor(Xtr_s); yb = torch.tensor(ytr, dtype=torch.long)
for epoch in range(5): #replace with 60
mlp.train()
opt.zero_grad()
loss = loss_fn(mlp(xb), yb)
loss.backward(); opt.step()
mlp.eval()
with torch.no_grad():
logits = mlp(torch.tensor(Xte_s))
pred = logits.argmax(1).numpy()
proba = torch.softmax(logits, dim=1)[:,1].numpy()
mlp_acc = accuracy_score(yte, pred)
mlp_auc = roc_auc_score(yte, proba)
print(f"LogReg baseline acc={base_acc_log:.3f} AUC={base_auc_log:.3f}")
print(f"RF baseline acc={base_acc_rf:.3f} AUC={base_auc_rf:.3f}")
print(f"MLP (scratch) acc={mlp_acc:.3f} AUC={mlp_auc:.3f}")