Lecture 3 - SMILES and RDKit#
Start with the SMILES language, practice tiny steps, then use RDKit to draw, edit, and analyze molecules. Finish with PubChem lookups.
Learning goals#
Read SMILES strings with confidence: atoms, bonds, branches, rings, aromaticity, charges, simple stereochemistry.
Use RDKit to parse SMILES, draw structures, add hydrogens, and compute basic properties.
Make small edits: replace atoms, neutralize groups, split salts, add a methyl group with a graph edit.
Query PubChem after you can edit molecules locally, then round-trip to SMILES and files.
 #
#
1. SMILES#
What is SMILES?
SMILES (Simplified Molecular Input Line Entry System) is a compact way to describe a molecule using only a line of text. It turns molecular structures into strings that are easy for both humans and computers to read.
Each atom is represented by its atomic symbol: C for carbon, O for oxygen, etc.
Bonds are shown with symbols: single bonds are implicit, = is a double bond, # is a triple bond.
Branches are placed in parentheses: CC(O)C means a side group on the middle carbon.
Ring structures use numbers to show where the ring closes: C1CCCCC1 is cyclohexane.
Charges, isotopes, and stereochemistry can also be encoded.
Because it’s text, SMILES is great for storing, comparing, and searching molecules in code and databases. RDKit can take a SMILES string, reconstruct the molecule, and let you visualize or analyze it.
For more information: J. Chem. Inf. Comput. Sci. 1988, 28 (1), 31–36
Now, let’s get started!
If you use Colab, run the install cell below first.
# Install only if needed
try:
import rdkit
from rdkit import Chem
from rdkit.Chem import Draw, Descriptors, Crippen, rdMolDescriptors
except Exception:
%pip install rdkit
1.1 Atoms#
Organic set without brackets:
B C N O P S F Cl Br I.Hydrogens are usually implicit.
Charges or unusual valence use brackets.
# Plain strings to focus on notation
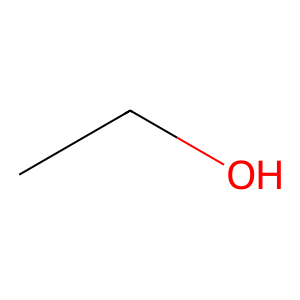
ethanol = "CCO" # C-C-O
mol_1 = Chem.MolFromSmiles(ethanol)
Draw.MolToImage(mol_1)
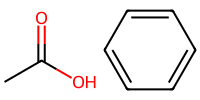
acetic = "CC(=O)O" # C-C with a double bonded O and an OH
benzene = "c1ccccc1" # aromatic ring
mol_2 = Chem.MolFromSmiles(acetic)
mol_3 = Chem.MolFromSmiles(benzene)
# Draw both molecules side by side
Draw.MolsToImage([mol_2, mol_3], molsPerRow=2, subImgSize=(100,100))
charged1 = "[NH4+]" # ammonium
charged2 = "C(=O)[O-]" # carboxylate
mol_c1 = Chem.MolFromSmiles(charged1)
mol_c2 = Chem.MolFromSmiles(charged2)
Draw.MolsToImage([mol_c1, mol_c2], molsPerRow=1, subImgSize=(100,100))
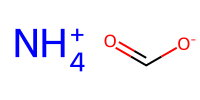
1.2 Bonds#
Single is implied.
=is double.#is triple.
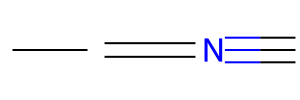
single = "CC"
double = "C=C"
triple = "C#N"
mols = [Chem.MolFromSmiles(bond) for bond in [single, double, triple]]
Draw.MolsToImage(mols, molsPerRow=1, subImgSize=(100,100))
1.3 Branches#
Parentheses create side branches.
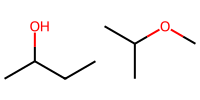
isopropanol_a = "CC(O)CC"
isopropanol_b = "CC(C)OC" # same structure, different order
print(isopropanol_a, isopropanol_b)
mol_a = Chem.MolFromSmiles(isopropanol_a)
mol_b = Chem.MolFromSmiles(isopropanol_b)
Draw.MolsToImage([mol_a, mol_b], molsPerRow=2, subImgSize=(100,100))
CC(O)CC CC(C)OC
1.4 Rings#
Numbers open and close rings.
Same digit appears twice to close that ring.
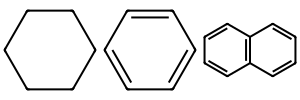
cyclohexane = "C1CCCCC1"
benzene = "c1ccccc1"
naphthalene = "c1cccc2c1cccc2"
mols = [Chem.MolFromSmiles(ring) for ring in [cyclohexane, benzene, naphthalene]]
Draw.MolsToImage(mols, molsPerRow=3, subImgSize=(100,100))
1.5 Aromatic vs aliphatic#
Aromatic atoms are lower case.
Aliphatic are upper case.
mol_aromatic = Chem.MolFromSmiles("c1ccccc1")
mol_aliphatic = Chem.MolFromSmiles("C1CCCCC1")
Draw.MolsToImage([ mol_aromatic, mol_aliphatic], molsPerRow=3, subImgSize=(100,100))
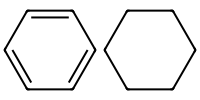
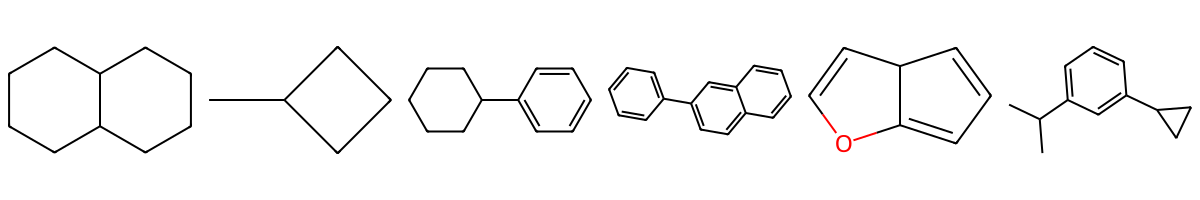
Note
What the digits mean: A number marks a ring connection. The same digit appears twice on the two atoms that are bonded to each other to close that ring. For fused systems, you can reuse different digits to show where each ring closes.
ring1 = Chem.MolFromSmiles("C1CCC2CCCCC2C1")
ring2 = Chem.MolFromSmiles("C1CC(C)C1")
ring3 = Chem.MolFromSmiles("c1ccc(C2CCCCC2)cc1")
ring4 = Chem.MolFromSmiles("c1ccc(-c2ccc3ccccc3c2)cc1")
ring5 = Chem.MolFromSmiles("C1=CC2C=COC2=C1")
ring6 = Chem.MolFromSmiles("CC(C)c1cccc(C2CC2)c1")
Draw.MolsToImage([ring1,ring2, ring3, ring4, ring5, ring6], ubImgSize=(100,100))
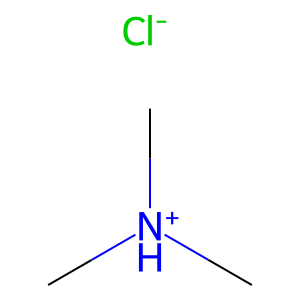
1.6 Charges and salts#
Use
.to separate parts in a salt.Place charge in brackets on the atom.
salt = "C[NH+](C)C.[Cl-]" # trimethylammonium chloride
Draw.MolToImage(Chem.MolFromSmiles(salt ), molsPerRow=1, subImgSize=(100,100))
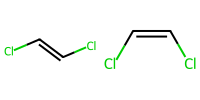
1.7 Simple stereochemistry#
E and Z for alkenes use slashes.
Cl/C=C/Clis E.Cl/C=C\Clis Z.
mol_E = Chem.MolFromSmiles("Cl/C=C/Cl")
mol_Z = Chem.MolFromSmiles("Cl/C=C\\Cl")
Draw.MolsToImage([ mol_E, mol_Z], molsPerRow=3, subImgSize=(100,100))
Try
Search online SMILES for these and print them:
isopropyl alcohol
benzoate anion
cyclopropane
pyridine
Try
Search online SMILES for these and print them:
isopropyl alcohol
benzoate anion
cyclopropane
pyridine
2. RDKit quick start#
from rdkit import Chem
from rdkit.Chem import Draw, Descriptors, Crippen, rdMolDescriptors
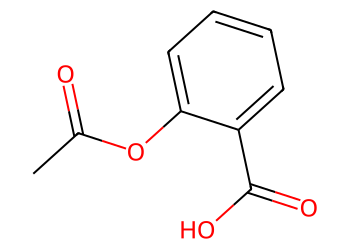
smi = "CC(=O)OC1=CC=CC=C1C(=O)O" # aspirin
mol = Chem.MolFromSmiles(smi)
Draw.MolToImage(mol, size=(350, 250))
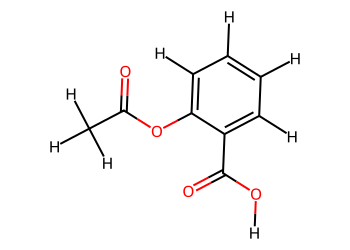
# Add hydrogens for clarity
mol_H = Chem.AddHs(mol)
Draw.MolToImage(mol_H, size=(350, 250))
# Quick properties in one place
mw = Descriptors.MolWt(mol)
logp = Crippen.MolLogP(mol)
hbd = rdMolDescriptors.CalcNumHBD(mol)
hba = rdMolDescriptors.CalcNumHBA(mol)
tpsa = rdMolDescriptors.CalcTPSA(mol)
print("MolWt", round(mw,2), "LogP", round(logp,2), "HBD", hbd, "HBA", hba, "TPSA", round(tpsa,1))
MolWt 180.16 LogP 1.31 HBD 1 HBA 3 TPSA 63.6
Note
MolWt → The molecular weight (molar mass) of the compound, measured in grams per mole.
LogP → The logarithm of the partition coefficient (octanol/water); higher values mean more lipophilic (hydrophobic).
HBD (Hydrogen Bond Donors) → Atoms (often OH or NH groups) that can donate a hydrogen in hydrogen bonding.
HBA (Hydrogen Bond Acceptors) → Atoms (such as oxygen or nitrogen) that can accept a hydrogen bond.
TPSA (Topological Polar Surface Area) → A measure of the molecule’s polar area, correlated with solubility and permeability.
# Show atom numbers to plan edits
img = Draw.MolToImage(mol, size=(350, 250), includeAtomNumbers=True)
img
Practice
Change smi to caffeine or acetaminophen. Compare MolWt and TPSA.
3. Small edits in RDKit#
We will avoid pattern languages here. We will use plain molecules to find and replace common pieces.
3.1 Replace atom type by matching a small molecule#
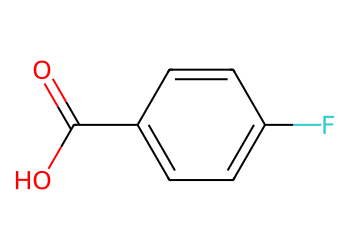
Replace all chlorine atoms with fluorine in an aryl chloride.
from rdkit import Chem
from rdkit.Chem import Draw
qry = Chem.MolFromSmiles("Cl") # what to find
rep = Chem.MolFromSmiles("F") # what to place
mol = Chem.MolFromSmiles("Clc1ccc(cc1)C(=O)O")
out = Chem.ReplaceSubstructs(mol, qry, rep, replaceAll=True)[0]
Draw.MolToImage(out, size=(350, 250))
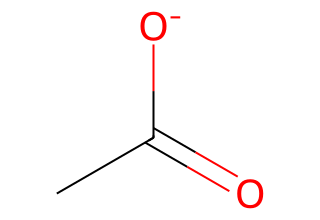
3.2 Neutralize a carboxylate#
mol = Chem.MolFromSmiles("CC(=O)[O-]")
find = Chem.MolFromSmiles("[O-]") # anionic oxygen as a molecule
put = Chem.MolFromSmiles("O")
mol_neutral = Chem.ReplaceSubstructs(mol, find, put, replaceAll=True)[0]
Draw.MolToImage(mol_neutral, size=(320, 220))
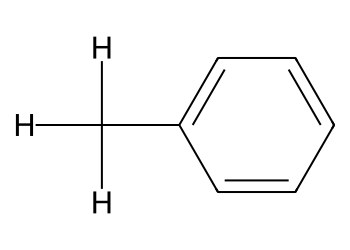
3.3 Add a methyl group with a graph edit#
mol = Chem.MolFromSmiles("c1ccccc1") # benzene
em = Chem.EditableMol(mol)
idx_C = em.AddAtom(Chem.Atom("C"))
idx_H1 = em.AddAtom(Chem.Atom("H"))
idx_H2 = em.AddAtom(Chem.Atom("H"))
idx_H3 = em.AddAtom(Chem.Atom("H"))
em.AddBond(2, idx_C, order=Chem.BondType.SINGLE) # attach at atom index 2
em.AddBond(idx_C, idx_H1, order=Chem.BondType.SINGLE)
em.AddBond(idx_C, idx_H2, order=Chem.BondType.SINGLE)
em.AddBond(idx_C, idx_H3, order=Chem.BondType.SINGLE)
mol2 = em.GetMol()
Chem.SanitizeMol(mol2)
Draw.MolToImage(mol2, size=(350, 250), includeAtomNumbers=True)
Tip
After graph edits, call Chem.SanitizeMol to check valence and aromaticity.
3.4 Why use EditableMol instead of just SMILES edits?#
Text edits to SMILES can hit the wrong atom or make an invalid string. EditableMol lets you target an atom index, keep valence correct, and apply the same change across many molecules in a repeatable way.
Now consider below example: add a methyl group to atom index 2 across 7 inputs
smiles_list = [
"c1ccccc1", # benzene
"Oc1ccccc1", # phenol
"Nc1ccccc1", # aniline
"Clc1ccccc1", # chlorobenzene
"c1ccncc1", # pyridine
"O=C(O)c1ccccc1", # benzoic acid
"OCCc1ccccc1", # benzyl alcohol
]
def add_methyl(smi):
mol = Chem.MolFromSmiles(smi) # parse SMILES to molecule
if mol is None or mol.GetNumAtoms() < 3: # quick guard for bad/short inputs
return None # signal failure
em = Chem.EditableMol(mol) # enter editable graph mode
c_idx = em.AddAtom(Chem.Atom("C")) # add the methyl carbon
for _ in range(3): # add three hydrogens
h = em.AddAtom(Chem.Atom("H")) # create a hydrogen atom
em.AddBond(c_idx, h, Chem.BondType.SINGLE) # connect H to C
em.AddBond(2, c_idx, Chem.BondType.SINGLE) # attach methyl C to atom index 2
newmol = em.GetMol() # exit edit mode
Chem.SanitizeMol(newmol) # check valence/aromaticity
return newmol # return edited molecule
for smi in smiles_list: # iterate over the 7 SMILES
mol2 = add_methyl(smi) # attempt the methyl add
out = Chem.MolToSmiles(mol2) if mol2 else "failed" # convert to output SMILES or flag
print("IN :", smi) # show input
print("---------------------")
print("OUT:", out) # show output
print("---------------------")
IN : c1ccccc1
---------------------
OUT: [H]C([H])([H])c1ccccc1
---------------------
IN : Oc1ccccc1
---------------------
OUT: [H]C([H])([H])c1ccccc1O
---------------------
IN : Nc1ccccc1
---------------------
OUT: [H]C([H])([H])c1ccccc1N
---------------------
IN : Clc1ccccc1
---------------------
OUT: [H]C([H])([H])c1ccccc1Cl
---------------------
IN : c1ccncc1
---------------------
OUT: [H]C([H])([H])c1ccccn1
---------------------
IN : O=C(O)c1ccccc1
---------------------
OUT: [H]C([H])([H])OC(=O)c1ccccc1
---------------------
IN : OCCc1ccccc1
---------------------
OUT: [H]C([H])([H])C(CO)c1ccccc1
---------------------
mols_in = [Chem.MolFromSmiles(s) for s in smiles_list] # make inputs as mols
mols_out = [add_methyl(s) for s in smiles_list] # make edited outputs
Draw.MolsToGridImage( # draw a grid to compare
mols_in + mols_out, # originals then edits
molsPerRow=5, # how many per row
subImgSize=(200,180), # image size
legends=["in"]*len(mols_in)+["out"]*len(mols_out), # labels
useSVG=True # SVG for crisp display
) # end drawing

4. Canonicalization and similarity#
4.1 Canonicalization#
SMILES strings are compact and easy to use, but there is a catch: they are not guaranteed to be unique.
The same molecule can be written with different SMILES depending on how the atoms are traversed. This makes it difficult to compare molecules directly.
In the example below we take three different SMILES notations, each of which corresponds to the molecule we have on quiz.
Although the strings differ, the structures are identical once interpreted by RDKit.
from rdkit import Chem
from rdkit.Chem import Draw
# Three equivalent SMILES
smile1 = "OCC=CCC(OC)CCC"
smile2 = "COC(CCC)CC=CCO"
smile3 = "CCCC(OC)CC=CCO"
smiles_list = [smile1, smile2, smile3]
mols = [Chem.MolFromSmiles(s) for s in smiles_list]
Draw.MolsToGridImage(mols, molsPerRow=3, subImgSize=(170,170),
legends=[f"form {i+1}" for i in range(len(mols))])
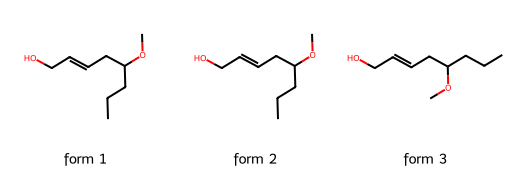
Note
Different SMILES strings can represent the same molecule. This is useful for flexibility, but problematic if we want a unique key for each compound.
Canonicalization generates a standard SMILES for each molecule, which allows unambiguous comparisons and avoids duplicates.
def canonicalize_smiles(smiles):
m = Chem.MolFromSmiles(smiles)
return Chem.MolToSmiles(m) if m is not None else None
for s in smiles_list:
print(s, " -> ", canonicalize_smiles(s))
OCC=CCC(OC)CCC -> CCCC(CC=CCO)OC
COC(CCC)CC=CCO -> CCCC(CC=CCO)OC
CCCC(OC)CC=CCO -> CCCC(CC=CCO)OC
Note
Canonicalization is not universal: RDKit and Open Babel may output different canonical strings for the same molecule.
The important part is consistency within your workflow—use the same toolkit throughout.
4.2 Morgan fingerprints and Tanimoto#
Another way to compare molecules is not by SMILES text but by molecular features.
Fingerprints encode the presence or absence of atom environments in a binary vector. Morgan fingerprints (circular fingerprints) are widely used.
So, what is a fingerprint?
Think of a fingerprint as a barcode for a molecule.
Each bar (or bit) in the barcode answers a yes/no question: Does the molecule contain this feature?
Example features: “Does it have a benzene ring?”, “Does it have an OH group?”, “Does it have a nitrogen atom with three bonds?”
If the answer is yes, the bit is 1. If not, it’s 0.
This gives us a long string of 0s and 1s that represents the molecule.
That’s said, Morgan fingerprints are a special kind of barcode. They look around each atom in the molecule and record the neighborhood (atoms directly attached, then atoms two steps away, etc.). The “radius” tells how far you look from each atom. Radius 2 means: “look two bonds away.” Morgan fingerprints are popular because they do a good job at capturing local environments around atom.
from rdkit.Chem import rdFingerprintGenerator
mfpgen = rdFingerprintGenerator.GetMorganGenerator(radius=2, fpSize=512)
methane = Chem.MolFromSmiles("C") # replace with other SMILES strings to see how it changes!
caffeine = Chem.MolFromSmiles("Cn1cnc2c1c(=O)n(C)c(=O)n2C")
fp_me = mfpgen.GetFingerprint(methane)
fp_caff = mfpgen.GetFingerprint(caffeine)
from rdkit import DataStructs
print(fp_me.ToBitString())
print("----------------")
print(fp_caff.ToBitString())
00000000000000000000000000000000000000000000000000000000000000000000000000000000000000000000000000000000000000000000000000000000000000000000000000000000000000000000000000000000000000000000000000000000000000000000000000000000000000000000000010000000000000000000000000000000000000000000000000000000000000000000000000000000000000000000000000000000000000000000000000000000000000000000000000000000000000000000000000000000000000000000000000000000000000000000000000000000000000000000000000000000000000000000000000000000
----------------
10000000000000000000000000000000010000000000000000001000000000000000000000000000000000000000000000000000000000000000000001000000000000000010000000000000000000001000000000000000000100000000000000000000000000000000000000000000000000000010000000000000000000000001000000000000000000000001000000000000000000000000000000100000000000000010000001000000000000000000100000000000000000000010000001000000000000001000000000000000100010010000100000000000000000000000000000000001000000000000000000000000000001000000000010000000
We can then measure how similar two fingerprints are with the Tanimoto coefficient.
It is one of the most common metrics for comparing chemical fingerprints and it measures how similar two molecules are by comparing their structural features represented as bit-vectors.
In terms of the formula, if we have two bit-vectors A and B:
a = number of bits set to 1 in A
b = number of bits set to 1 in B
c = number of common bits set to 1 in both A and B
Then the Tanimoto similarity is:
The numerator c counts the shared features (substructures present in both molecules).
The denominator counts all unique features across both molecules, subtracting the overlap.
The result ranges between 0 and 1:
0→ no similarity at all.1→ identical fingerprints.
Suppose we compare caffeine and theobromine with Morgan fingerprints.
Caffeine has about 250 “on” bits (features).
Theobromine has about 240 “on” bits.
They share about 180 bits.
This means caffeine and theobromine share 58% of their features.
By contrast, comparing caffeine with phenol may yield ~0.15, showing much weaker similarity.
Note
Tanimoto is equivalent to the Jaccard index used in other fields.
Other similarity measures exist (Dice, Cosine, Hamming), but Tanimoto remains the standard in cheminformatics.
Choice of fingerprint (Morgan, MACCS, topological) influences the score.
from rdkit.Chem import rdFingerprintGenerator
from rdkit.DataStructs import FingerprintSimilarity
caffeine = Chem.MolFromSmiles("Cn1cnc2c1c(=O)n(C)c(=O)n2C")
theobromine = Chem.MolFromSmiles("Cn1c(=O)c2[nH]c(nc2n(C)c1=O)C")
phenol = Chem.MolFromSmiles("c1ccccc1O")
mfpgen = rdFingerprintGenerator.GetMorganGenerator(radius=2, fpSize=1024)
fp_caff = mfpgen.GetFingerprint(caffeine)
fp_theo = mfpgen.GetFingerprint(theobromine)
fp_phenol = mfpgen.GetFingerprint(phenol)
print("Caffeine vs theobromine:", round(FingerprintSimilarity(fp_caff, fp_theo),3))
print("Caffeine vs phenol :", round(FingerprintSimilarity(fp_caff, fp_phenol),3))
Caffeine vs theobromine: 0.389
Caffeine vs phenol : 0.061
Note
Notice how caffeine and theobromine have a high similarity because they share a xanthine core.
Phenol is much less similar since it only shares a benzene ring fragment with caffeine.
4.3 Bemis–Murcko scaffold#
To simplify molecules further we can extract their scaffolds, which keep the core ring system and linkers but discard side chains.
from rdkit.Chem.Scaffolds import MurckoScaffold
mol_carboxy = Chem.MolFromSmiles("O=C(O)c1ccc(C(=O)O)cc1")
mol_hydroxamate = Chem.MolFromSmiles("O=C(O)c1ccc(C(=O)NO)cc1")
scaf_c = MurckoScaffold.GetScaffoldForMol(mol_carboxy )
scaf_h = MurckoScaffold.GetScaffoldForMol(mol_hydroxamate)
Draw.MolsToGridImage([mol_carboxy, scaf_c , mol_hydroxamate, scaf_h ],
legends=["mol_c", "scaffold_c", "mol_hydroxamate", "scaffold_h"],
molsPerRow=2, subImgSize=(180,180))
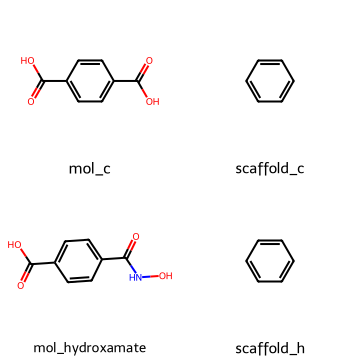
Both of the two molecules above share the same benzene scaffold substructure.
from rdkit.Chem.Scaffolds import MurckoScaffold
scaf_caff = MurckoScaffold.GetScaffoldForMol(caffeine)
scaf_theo = MurckoScaffold.GetScaffoldForMol(theobromine)
Draw.MolsToGridImage([caffeine, scaf_caff, theobromine, scaf_theo],
legends=["caffeine", "caffeine scaffold", "theobromine", "theobromine scaffold"],
molsPerRow=2, subImgSize=(180,180))
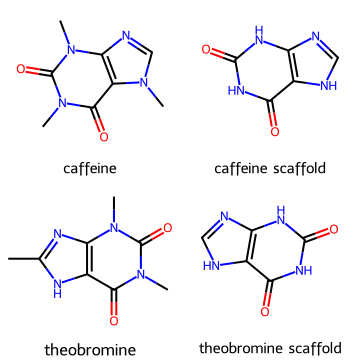
Note
Scaffolds are useful in medicinal chemistry to group compounds into families.
Different analogs may have the same core scaffold but different substituents that tune activity or solubility.
5. RDKit with pandas#
Large datasets are often stored as tables. Pandas can manage these datasets, while RDKit enriches them with molecular objects and descriptors.
5.1 Load dataset and add molecule column#
import pandas as pd
from rdkit.Chem import PandasTools
url = "https://raw.githubusercontent.com/zzhenglab/ai4chem/main/book/_data/organic_ligands_inventory_smiles.xlsx"
df = pd.read_excel(url, engine="openpyxl")
df = df[['Compound ID','smiles','MW']]
PandasTools.AddMoleculeColumnToFrame(df, smilesCol='smiles', molCol='Mol')
df.head()
| Compound ID | smiles | MW | Mol | |
|---|---|---|---|---|
| 0 | 2-Methylimidazole | CC1=NC=CN1 | 82.106 | <rdkit.Chem.rdchem.Mol object at 0x000002235D7... |
| 1 | 1-Ethyl-3-vinylimidazolium bromide | C=C[N+]1=CN(CC)C=C1.[Br-] | 203.083 | <rdkit.Chem.rdchem.Mol object at 0x000002235D7... |
| 2 | 1-Methyl-1,4-diazabicyclo[2.2.2]octan-1-iumchl... | [Cl-].N12CC[N+](C)(CC1)CC2 | 162.660 | <rdkit.Chem.rdchem.Mol object at 0x000002235D7... |
| 3 | Oxazole-4-carboxylic acid | O=C(C1=COC=N1)O | 113.072 | <rdkit.Chem.rdchem.Mol object at 0x000002235D7... |
| 4 | 3-Ethyl-2-methylbenzo[d]thiazol-3-ium iodide | CC[N+]1=C(C)SC2=CC=CC=C21.[I-] | 305.184 | <rdkit.Chem.rdchem.Mol object at 0x000002235D7... |
Note
Adding a Mol column converts SMILES to RDKit molecules directly in the DataFrame.
This makes it easy to compute descriptors row by row or draw molecules inline.
5.2 Canonicalize SMILES and compute descriptors#
def canonicalize(smiles):
m = Chem.MolFromSmiles(smiles)
return Chem.MolToSmiles(m) if m else None
df['canonical_smiles'] = df['smiles'].apply(canonicalize)
Now we create a new collum ‘canonical_smiles’ with the canonicalized smiles.
from rdkit.ML.Descriptors.MoleculeDescriptors import MolecularDescriptorCalculator
desc_names = ['MolWt','TPSA','MolLogP','NumHAcceptors','NumHDonors']
calc = MolecularDescriptorCalculator(desc_names)
df_desc = df['Mol'].apply(calc.CalcDescriptors)
df_desc = pd.DataFrame(df_desc.tolist(), columns=desc_names)
df_all = pd.concat([df[['Compound ID','MW','canonical_smiles']], df_desc], axis=1)
df_all.head()
| Compound ID | MW | canonical_smiles | MolWt | TPSA | MolLogP | NumHAcceptors | NumHDonors | |
|---|---|---|---|---|---|---|---|---|
| 0 | 2-Methylimidazole | 82.106 | Cc1ncc[nH]1 | 82.106 | 28.68 | 0.71812 | 1 | 1 |
| 1 | 1-Ethyl-3-vinylimidazolium bromide | 203.083 | C=C[n+]1ccn(CC)c1.[Br-] | 203.083 | 8.81 | -2.10000 | 1 | 0 |
| 2 | 1-Methyl-1,4-diazabicyclo[2.2.2]octan-1-iumchl... | 162.660 | C[N+]12CCN(CC1)CC2.[Cl-] | 162.664 | 3.24 | -3.23380 | 1 | 0 |
| 3 | Oxazole-4-carboxylic acid | 113.072 | O=C(O)c1cocn1 | 113.072 | 63.33 | 0.37280 | 3 | 1 |
| 4 | 3-Ethyl-2-methylbenzo[d]thiazol-3-ium iodide | 305.184 | CC[n+]1c(C)sc2ccccc21.[I-] | 305.184 | 3.88 | -0.47888 | 1 | 0 |
Note
Descriptors such as TPSA and LogP are common in QSAR modeling and drug-likeness rules.
For example, Lipinski’s rule-of-five uses MolWt, LogP, HBD, and HBA thresholds.
5.3 Clean and visualize#
df_clean = df_all.dropna()
print("Number of rows after cleaning:", len(df_clean))
Number of rows after cleaning: 102
import matplotlib.pyplot as plt
plt.hist(df_clean['MolWt'], bins=40)
plt.xlabel("Molecular Weight (g/mol)")
plt.ylabel("Count")
plt.show()
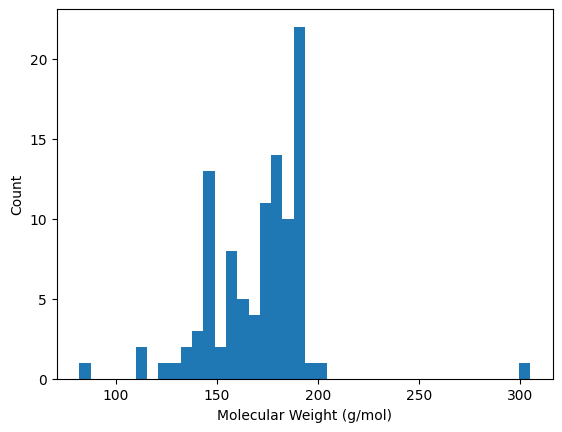
Note
Cleaning removes molecules that failed parsing. Histograms allow you to inspect the distribution of properties—are most molecules small and drug-like, or large and polar?
6. Quick reference#
SMILES
Atoms: upper case aliphatic, lower case aromatic
Bonds: implicit single, =, #
Branches: parentheses
Rings: digits to open and close
Charges: bracket the atom, e.g., [O-], [NH4+]
Salts: separate parts with a dot
E or Z: use slashes around the double bond
RDKit
Parse:
Chem.MolFromSmilesDraw:
Draw.MolToImage(..., includeAtomNumbers=True)Hydrogens:
Chem.AddHsProperties:
Descriptors.MolWt,Crippen.MolLogP,CalcNumHBA/HBD,CalcTPSAReplace piece with piece:
Chem.ReplaceSubstructs(mol, findMol, repMol)Graph edit:
Chem.EditableMolSave:
Chem.MolToSmiles,SDWriter, PNG viaMolToImage(...).save(...)
7. Glossary#
- SMILES#
Text line notation for molecules. Example: ethanol is CCO.
- aromatic#
Conjugated ring system shown with lower case atom symbols in SMILES, for example c1ccccc1.
- CID#
PubChem Compound ID for a unique compound record.
- sanitize#
RDKit process that checks valence, aromaticity, and stereochemistry.
- descriptor#
Computed molecular property such as molecular weight or LogP.
- EditableMol#
RDKit object that exposes low level atom and bond editing.
- Bemis–Murcko scaffold#
The ring system and linkers that form a molecule’s core framework.
8. In-class activity#
Each task mirrors the examples above. Fill in the ... lines. Work in pairs. Solutions are in Section 9.
8.1 Read a SMILES and inspect#
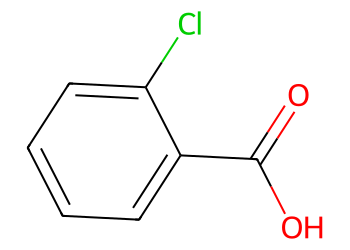
Given smi = "O=C(O)c1ccccc1Cl".
a) Count number of rings.
bc) Print the list of bonds with begin and end atom indices and bond orders.
from rdkit import Chem
from rdkit.Chem import Draw
smi = ... # TO DO
mol = Chem.MolFromSmiles(smi)
display(Draw.MolToImage(mol, size=(350, 250), includeAtomNumbers=True))
num_rings = ... # TO DO: Chem.GetSSSR(mol)
print("rings:", num_rings)
for b in mol.GetBonds():
print("bond", b.GetIdx(), b.GetBeginAtomIdx(), "-", b.GetEndAtomIdx(), "order", int(b.GetBondTypeAsDouble()))
8.2 Make a small properties table#
Use smiles ["Cn1cnc2N(C)C(=O)N(C)C(=O)c12", "CC(=O)Nc1ccc(O)cc1", "CC(C)Cc1ccc(cc1)C(C)C(O)=O"]. For each, compute MolWt, LogP, HBD, HBA, and TPSA.
import pandas as pd
from rdkit.Chem import Descriptors, Crippen, rdMolDescriptors
names = ... # TO DO
rows = []
for nm in names:
... #TO DO
rows.append({
"smiles": ...,
"MolWt": ...,
"LogP": ...,
"HBD": ...,
"HBA": ...,
"TPSA": ...
})
pd.DataFrame(rows)
8.3 Replace chlorine with fluorine#
Replace Cl with F in Clc1ccc(cc1)C(=O)O and print the result SMILES.
find = Chem.MolFromSmiles(... ) # TO DO
put = Chem.MolFromSmiles(... ) # TO DO
mol = ... # TO DO
out = ... # TO DO
print(Chem.MolToSmiles(out))
8.4 Add a methyl group with a graph edit#
Add a methyl at atom index 2 of benzene.
mol = Chem.MolFromSmiles("c1ccccc1")
em = Chem.EditableMol(mol)
# TO DO: add code
Draw.MolToImage(..., size=(350, 250))
8.5 Retrieve SMILES from PubChem Draw structure, then analyze#
Images to inspect:
Show code cell source
from IPython.display import Image, display
# Show provided images for reference
display(Image(url="https://raw.githubusercontent.com/zzhenglab/ai4chem/main/book/_data/lec-3-structures.png"))
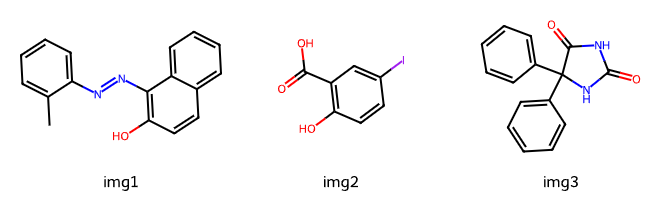
Click Draw structure.
Paste draw the structure above, then copy the SMILES.
Use the SMILES in the cell below to draw with RDKit and compute basic properties.
# Paste the SMILES you obtained from PubChem Draw structure
smi1 = "..." # smiles for image 1
smi2 = "..." # smiles for image 2
smi3 = "..." # smiles for image 3
m1 = Chem.MolFromSmiles(smi1)
m2 = Chem.MolFromSmiles(smi2)
m3 = Chem.MolFromSmiles(smi3)
Draw.MolsToGridImage([m1, m2, m3], legends=["img1","img2","img3"], molsPerRow=3, subImgSize=(220,200), useSVG=True)
# Compute quick properties for the three molecules
from rdkit.Chem import Descriptors, Crippen, rdMolDescriptors
import pandas as pd
... # TO DO: get MolWt=..., LogP=...,HBD=...,HBA=..., TPSA=...
df = pd.DataFrame([
{"name":"img1","smiles":smi1, ...},
{"name":"img2","smiles":smi2, ...},
{"name":"img3","smiles":smi3, ...}
]).round(3)
df
9. Solutions#
Open after you try Section 8.
Solution 8.1#
from rdkit import Chem
from rdkit.Chem import Draw
smi = "O=C(O)c1ccccc1Cl"
mol = Chem.MolFromSmiles(smi)
display(Draw.MolToImage(mol, size=(350, 250), includeAtomNumbers=True))
num_rings = Chem.GetSSSR(mol)
print("rings:", num_rings)
for b in mol.GetBonds():
print("bond", b.GetIdx(), b.GetBeginAtomIdx(), "-", b.GetEndAtomIdx(), "order", int(b.GetBondTypeAsDouble()))
rings: <rdkit.rdBase._vectclass std::vector<int,class std::allocator<int> > object at 0x000002235E4ED740>
bond 0 0 - 1 order 2
bond 1 1 - 2 order 1
bond 2 1 - 3 order 1
bond 3 3 - 4 order 1
bond 4 4 - 5 order 1
bond 5 5 - 6 order 1
bond 6 6 - 7 order 1
bond 7 7 - 8 order 1
bond 8 8 - 9 order 1
bond 9 8 - 3 order 1
Solution 8.2#
import pandas as pd
from rdkit.Chem import Descriptors, Crippen, rdMolDescriptors
names = ["Cn1cnc2N(C)C(=O)N(C)C(=O)c12", "CC(=O)Nc1ccc(O)cc1", "CC(C)Cc1ccc(cc1)C(C)C(O)=O"]
rows = []
for nm in names:
m = Chem.MolFromSmiles(nm)
rows.append({
"smiles": nm,
"MolWt": Descriptors.MolWt(m),
"LogP": Crippen.MolLogP(m),
"HBD": rdMolDescriptors.CalcNumHBD(m),
"HBA": rdMolDescriptors.CalcNumHBA(m),
"TPSA": rdMolDescriptors.CalcTPSA(m)
})
pd.DataFrame(rows)
| smiles | MolWt | LogP | HBD | HBA | TPSA | |
|---|---|---|---|---|---|---|
| 0 | Cn1cnc2N(C)C(=O)N(C)C(=O)c12 | 194.194 | -1.0293 | 0 | 6 | 61.82 |
| 1 | CC(=O)Nc1ccc(O)cc1 | 151.165 | 1.3506 | 2 | 2 | 49.33 |
| 2 | CC(C)Cc1ccc(cc1)C(C)C(O)=O | 206.285 | 3.0732 | 1 | 1 | 37.30 |
Solution 8.3#
find = Chem.MolFromSmiles("Cl")
put = Chem.MolFromSmiles("F")
mol = Chem.MolFromSmiles("Clc1ccc(cc1)C(=O)O")
out = Chem.ReplaceSubstructs(mol, find, put, replaceAll=True)[0]
print(Chem.MolToSmiles(out))
Draw.MolToImage(out, size=(350, 250))
O=C(O)c1ccc(F)cc1
Solution 8.4#
mol = Chem.MolFromSmiles("c1ccccc1")
em = Chem.EditableMol(mol)
idx_C = em.AddAtom(Chem.Atom("C"))
idx_H1 = em.AddAtom(Chem.Atom("H"))
idx_H2 = em.AddAtom(Chem.Atom("H"))
idx_H3 = em.AddAtom(Chem.Atom("H"))
em.AddBond(2, idx_C, order=Chem.BondType.SINGLE)
em.AddBond(idx_C, idx_H1, order=Chem.BondType.SINGLE)
em.AddBond(idx_C, idx_H2, order=Chem.BondType.SINGLE)
em.AddBond(idx_C, idx_H3, order=Chem.BondType.SINGLE)
mol2 = em.GetMol()
Chem.SanitizeMol(mol2)
Draw.MolToImage(mol2, size=(350, 250), includeAtomNumbers=True)